


Rhabdomyosarcome
Qu'est-Ce Qu'un Rhabdomyosarcome?
Le corps humain comprend 2 types de cellules musculaires différents: celles des muscles lisses et celles des muscles squelettiques. Les muscles lisses contrôlent les activités involontaires tandis que les muscles squelettiques contrôlent les activités volontaires. Les rhabdomyosarcomes (RMS) sont des tumeurs malignes ("cancer") qui se développent à partir du tissu musculaire squelettique normal. Les raisons de cette transformation d'une cellule normale en cellule cancéreuse restent à être élucidées. Puisque l'on trouve du tissu musculaire squelettique dans l'ensemble du corps humain, un rhabdomyosarcome peut se développer presque dans n'importe quelle partie du corps humain.
Les RMS furent décrits pour la première fois par Weber en 1854. Cependant, la publication "définitive" est généralement considérée ayant été élaborée par Stout en 1946, 92 ans plus tard.
- Weber, CO. Anatomische Untersuchung Einer Hypertrophieschen Zunge nebst Bemekugen uber die Nubildung querquestreifter Muskelfsern, Virchow Arch. Pathol Anat. 7:115, 1854.
- Stout AP: Rhabdomyosarcoma of the skeletal muscles, Ann Surg 1946; 123: 447-472.

Le RMS est un cancer très rare. Seulement 350 nouveaux cas de RMS sont diagnostiqués chaque année aux Etats-Unis chez les jeunes de moins de 21 ans. Environ 4 enfants pour un million d'enfants de moins de 15 ans en bonne santé développent un RMS chaque année. Ce cancer touche les garçons légèrement plus que les filles et est plus fréquent chez les jeunes enfants de moins de 5 ans.
Les RMS sont rares chez l'adulte. 5 études avec un large nombre de patients ont été publiées comprenant un total de 400 cas d'adultes atteints de RMS (incluant quelques "enfants") soignés dans des centres experts parmi les plus grands, aux Etats-Unis et en Europe, dans les 20 à 30 dernières années (voir références 1 à 5). Bien que l'histologie pléomorphe soit plus commune parmi la population adulte (et rarement vue chez l'enfant), les modalités de traitement pour la prise en charge des adultes atteints de RMS sont similaires à celles mises en place chez les enfants et leur évolution n'est intrinsèquement pas plus mauvaise chez les adultes traités par une thérapie "moderne" multidisciplinaire.
Cas Adultes
 Les modalités de traitement pour la prise en charge des adultes atteints de RMS sont similaires à celles des enfants. Les 5 publications énoncées précédemment proviennent de:
Les modalités de traitement pour la prise en charge des adultes atteints de RMS sont similaires à celles des enfants. Les 5 publications énoncées précédemment proviennent de:
- Instituto Nazionale Tumori, Milan, Italie, 190 patients âgés de 18 ans ou plus sur une période de 25 ans (réf. 1)
- Memorial Sloan-Kettering Cancer Center, New York City, NY, 84 patients âgés de 16 ans ou plus sur une période de 17 ans (réf. 2)
- M.D. Anderson Cancer Center, Houston, TX, 82 patients âgés de 17 ans ou plus sur une période de 28 ans (réf 3)
- Dana-Farber Cancer Institute, Boston, MA, 39 patients âgés de 16 ans ou plus sur une période de 23 ans (réf 4)
- Armed Forces Institute of Pathology (Washington, D.C., 38 patients âgés de 21 ans ou plus sur une période de 30 ans, tous atteints de RMS pléomorphes (réf 5)
Ils soulignent plusieurs points clés à propos des RMS de l’adultes:
- Ceux-ci répondent intrinsèquement aussi bien à la chimiothérapie que les RMS pédiatriques, avec des taux de réponse pouvant atteindre 85%
- Les histologies "défavorables", qui comprennent les sous-types alvéolaires et pléomorphes sont plus communes que celle du sous-type embryonnaire
- La proportion de patients ayant des tumeurs du groupe I, II, III et IV est comparable à celle observée dans les études pédiatriques
- Après un traitement adapté, même en considérant la différence de proportion en terme de patients aux histologies "défavorables", des taux de survie comparables à ceux observés dans les études pédiatriques peuvent être atteints
Bien que ces tumeurs peuvent apparaitre presque n'importe où, les emplacements les plus fréquentes du développement de ces tumeurs sont au niveau des structures de la tête et du cou (environ 40% de l'ensemble des cas), au niveau de l'appareil génito-urinaire masculin ou féminin (environ 25% de tous les cas) et au niveau des membres (environ 20% des cas).
Localisation |
% |
|---|---|
Para-méningée |
16 |
Orbite |
10 |
Tête/cou |
10 |
Génito-urinaire |
23 |
Membre |
19 |
Autre |
22 |
Environ 40% des nouveaux cas de RMS surviennent au niveau de la tête et du cou, ce qui incluent les localisations para-méningées (16% de tous les cas et presque la moitié des cas au niveau de la tête et du cou), l'orbite ou les paupières (10% de tous les cas) et autres localisations (10% de tous les cas). Environ 25% des cas surviennent au niveau de l'appareil génito-urinaire qui inclut la localisation para-testiculaire, l'appareil génito-urinaire féminin (vulve, vagin, col de l'utérus, utérus), la vessie et la prostate. Environ 20% des cas surviennent dans un membre. Le reste des cas surviennent dans des emplacements divers dont la cavité thoracique et le rétropéritoine.
Les tumeurs qui se présentent au niveau de l'orbite, des localisations non para-méningées de la tête et du cou (par exemple la joue ou le lobe de l'oreille) et des appareils génito-urinaires masculins (paratesticulaire) ou féminins (vagin, vulve, col de l'utérus ou utérus), sont considérées "favorables". Tous les autres emplacements sont considérés comme "défavorables".
La plupart des enfants atteints de RMS n'ont pas de facteur de risque connu pour développer un cancer. Après une étude soigneuse de l'historique familial et un examen physique complet, de 1 à 5 enfants sur 10 présentera un facteur de risque génétique identifiable. Les "syndromes" génétiques les plus courants sont : le syndrome de Li-Fraumeni (réf. 6), la neurofibromatose (réf. 7), le syndrome de Beckwith-Wiedemann (réf. 8) et le syndrome de Costello (réf. 9).
Bien que la grande majorité des cas de RMS se présentent de façon sporadique, entre 10 à 33% des enfants atteints de RMS semblent présenter un facteur de risque génétique sous-jacent (réf. 10). Le développement des RMS est associé à un certain nombre de syndromes familiaux rares liés au cancer tels que le syndrome de Li-Fraumeni qui est associé à un regroupement au sein d'une famille de RMS ou autres sarcomes des tissus mous chez les enfants et carcinomes adrénocorticaux ou carcinomes précoces du sein parmi les adultes. Le syndrome de Li-Fraumeni est associé à une mutation germinale du gène p53 (réf. 12). Les RMS apparaissent également en association avec le syndrome de Beckwith-Wiedemann caractérisé par une croissance excessive du fœtus associée à des anomalies dans la région 11p15 où se trouve le gène du facteur de croissance-2 analogue à l'insuline (IGF-2). Des études sur les enfants atteints du syndrome de Costello, vraisemblablement de transmission autosomique dominante, caractérisé par un retard de croissance post-natal, des traits épais, une peau relâchée et un retard au développement, révèlent un risque accru de développement de tumeurs solides, le plus couramment de RMS. 10 cas de RMS ont été signalés pour environ 100 cas d'enfants atteints du syndrome de Costello.
Los estudios de niños con síndrome de Costello, un desorden autosómico dominante caracterizado por retardo del crecimiento post-natal, rasgos faciales toscos típicos, piel laxa y retraso del desarrollo, han demostrado un riesgo incrementado de desarrollo de tumores sólidos, más frecuentemente rabdomiosarcomas. Se han descrito 10 casos de RMS en el grupo de unos 100 casos conocidos de niños con síndrome de Costello.
Symptômes des RMS
Les symptômes associés aux RMS sont très variables selon la localisation de la tumeur. Les enfants atteints d'un RMS orbitaire (environ 10% de tous les cas de RMS) peuvent présenter un oeil gonflé ou enflé (proptose / Exophtalmie). Bien que cela puisse parfois être confondu avec une infection des sinus, les enfants atteints de ces tumeurs n'ont généralement pas les autres symptômes des enfants présentant une infection des sinus (douleur, fièvre, décoloration violette de l'oeil).
Cas 1

Un garçon âgé de 7 ans se présente avec une douleur et un gonflement de l'oeil gauche qui durent depuis une semaine, sans fièvre ni rhinorrhée purulente. Des antibiotiques sont administrés par intraveineuse pour le traitement d'une cellulite péri-orbitaire présumée. Une IRM (figure 2) est obtenue et révèle une masse de tissu mou d'environ 4 cm, se développant au niveau de l'angle supéro-médial de l'orbite gauche, déplaçant le globe oculaire en avant et latéralement. Une biopsie de la masse est obtenue par une petite incision réalisée médialement. Le diagnostic d'un RMS embryonnaire est ainsi confirmé. Aucune métastase distante n'est trouvée par scanner thoracique, scintigraphie osseuse ou par biopsie de la moelle osseuse. Le patient est classé en stade 1, groupe III et est traité avec succès par chimiothérapie VA (vincristine/actinomycin) et radiothérapie locale (45 Gy).
Les enfants dont les tumeurs se développent dans une des localisations paraméningées (fondamentalement les sinus, l'oreille moyenne et le fond de la gorge) peuvent se plaindre pendant des semaines ou des mois d'avoir le nez bouché avec parfois un écoulement nasal. A l'occasion, une masse peut être visible dans une narine ou au fond de la gorge. Contrairement aux infections des sinus et de la gorge, ces tumeurs ne se propagent généralement pas dans les ganglions lymphatiques du cou. Si cela est le cas, les ganglions ne sont généralement pas sensibles au toucher. Si la base du crâne se trouve endommagée, le patient peut se plaindre de maux de tête et développer des neuropathies crâniennes dues à l'infiltration ou compression des nerfs crâniens affectés.
Cas 2

Une jeune fille de 14 ans se présente avec un historique de proptose du côté droit s'aggravant rapidement sur une période de 2 semaines et des ganglions gonflés du côté droit du cou. L'IRM montre une masse du tissu mou à compartiments multiples de presque 7 cm (figure 3) centrée sur la cavité naso-sinusienne s'étendant au travers de la lame criblée jusqu'à la fosse crânienne antérieure. Aucun œdème n'est présent au niveau des lobes frontaux qui puisse suggérer une directe extension parenchymateuse de la tumeur. Plusieurs ganglions lymphatiques gonflés sont également présents dans la région rétropharyngée latérale droite et dans la chaîne cervicale antérieure droite. L'examen physique met en évidence une proptose marquée du côté droit et une ophtalmoplégie sans perte de vision. Une masse est visible dans la narine droite. Une adénopathie cervicale aussi dure qu'une pierre est présente. Une ponction à l'aiguille fine (PAF) des ganglions cervicaux révèle une petite tumeur à cellules rondes bleues faisant suspecter un RMS. Une biopsie de la masse de la cavité nasale montre l'apparence "alvéolaire" caractéristique des RMS alvéolaires. Les immunomarquages sont fortement positifs pour la desmine, vimentine et la myogénine. La RT-PCR confirme la présence de la translocation t(2;13) PAX3-FKHR. La cytologie du liquide céphalorachidien ne détecte pas de cellule maligne. Aucune métastase distante n'est détectée par scanner thoracique, scintigraphie osseuse ou biopsie de la moelle épinière. Le diagnostic conclut un RMS alvéolaire de stade 3, Groupe III d'origine paraméningée (probablement au niveau de l'ethmoïde) avec extension intracrânienne. Suite à deux cycles de chimiothérapie, l'IRM et le PET-scan révèlent la disparition de la tumeur visible au préalable. Malgré l'administration additionnelle de chimiothérapie et une radiothérapie à dose complète (50.4 Gy) au niveau de l'emplacement d'origine et de tous les ganglions atteints, une rechute au niveau des leptoméninges progressant rapidement et éventuellement fatale est décrite, dans le champ irradié, 6 mois après le début du traitement.
Les enfants atteints de tumeurs se développant au niveau de l'appareil génito-urinaire peuvent présenter une masse scrotale indolore (tumeurs paratesticulaires), une masse en forme de grappe au niveau du vagin (RMS "botryoïde"), du sang dans les urines (tumeurs de la vessie), ou une urination fréquente, avec parfois brûlure ou hésitation. Occasionnellement, les tumeurs qui se développent au niveau de la prostate (différent du type de cancer de la prostate chez les hommes adultes) peuvent devenir très larges avant d'être diagnostiquées. Ces tumeurs peuvent se présenter comme une masse visible du pelvis ou de l'abdomen, parfois avec pollakiurie et miction impérieuse, parfois avec constipation, nausées et vomissements dus à la compression des intestins.
Cas 3

Un étudiant de 18 ans se présente avec une dysfonction érectile, des douleurs abdominales aiguës, des douleurs lombaires du côté droit, de la pollakiurie, une miction impérieuse et une diminution du débit urinaire. L'administration d'antibiotiques oraux ne génère aucune amélioration. Un scanner montre une masse pelvienne de 10 x 6.5 x 7.3 cm se développant à proximité de la prostate, inséparable de la paroi postérieure de la vessie et de la paroi antérieure du rectum, obstruant l'uretère droit, causant une hydronéphrose droite, avec adénopathie iliaque externe bilatérale et interne à gauche. L'IRM donne des résultats similaires (figure 4). Une biopsie transrectale à l'aiguille révèle un tissu comprenant une tumeur de forte densité cellulaire avec des petites cellules rondes bleues, fortement positive à la desmine, vimentine, actine, et myogénine lors de l'immunomarquage et contenant la translocation t(2;13) PAX3-FKHR d'après la RT-PCR. Un tube de néphrostomie percutanée est temporairement inséré pour soulager l'hydrophénose droite. Aucune métastase distante n'est détectée par scanner thoracique, scintigraphie osseuse ou biopsie de la moelle épinière. Le diagnostic conclut à un RMS alvéolaire de la prostate de stade 3, groupe III . Une chimiothérapie multi-agents, agressive, est administrée et une réponse complète en résulte. La fonction érectile redevient normale. Une chimiothérapie complémentaire et une radiothérapie du pelvis à dose complète (50.4 Gy) sont également administrées. Le traitement est compliqué par le développement d'une cystite hémorragique et d'une entérite radique. Le patient retourne à ses études moins de trois mois après la fin de son traitement de 8 mois et est toujours en rémission complète 18 mois après son diagnostic.
Les tumeurs qui se développent dans les jambes ou les bras font généralement partie des types de RMS les plus agressifs. Ces tumeurs peuvent passer de la taille d'une piqûre de moustique ou d'une petite bille à la taille d'une balle de baseball ou d'un pamplemousse en seulement quelques semaines. Ces tumeurs sont généralement dures au toucher mais sont rarement douloureuses à moins qu'elles ne compressent un nerf adjacent. Ces tumeurs sont les plus susceptibles à atteindre les ganglions adjacents. Les enfants atteints d'un RMS à la main ou au bras ont souvent les ganglions de l'aisselle gonflés et ceux atteints d'un RMS au pied ou au mollet ont les ganglions de l'aine gonflés.
Cas 4

Un garçon de 7 ans se présente suite à la découverte au cours d'un bain d'une masse ferme, indolore au niveau du mollet gauche. Un examen physique confirme la présence de la masse dure, comme de la pierre, au niveau du mollet avec des ganglions poplités et inguinaux manifestement gonflés. L'IRM (figure 5) montre une masse de tissu mou large dans le mollet avec présence d'hémorragie, qui s'étend antérieurement au travers de la fosse poplitée. Un scanner du thorax, abdomen et pelvis révèle la présence d'adénopathie inguinale et pelvienne et d'une adénopathie para-aortique "suspecte". Un PET scan confirme l'hypermétabolisme de ces ganglions cohérent avec la présence de métastases. Une biopsie incisionnelle de la masse du mollet et du ganglion inguinal conclut à un RMS "classique" alvéolaire. La RT-PCR confirme la présence de la translocation PAX-FKHR. Mis à part pour les métastases ganglionnaires, aucune autre métastase n'est détectée dans les poumons, les os ou la moelle épinière. Le diagnostic conclut à un RMS alvéolaire des membres de stade 4, group IV avec métastases ganglionnaires régionales (poplitées et inguinales) et distantes (pelviennes et para-aortiques). Durant la semaine suivant le début de la chimiothérapie, la tumeur du mollet régresse de plus de 50% et les ganglions ne sont plus hypermétaboliques. Le traitement continue suivant un protocole pilote du Memorial Sloan Kettering Cancer Center pour patients à "risque élevé".
Occasionnellement, les enfants atteints de RMS souffrent également de fièvres inexpliquées qui sont l'un des symptômes remarquables au moment du diagnostic. L'appétit peut ou pas être diminué. La fatigue et les contusions faciles sont rares à moins que la tumeur ne se soit étendue à la moelle osseuse.
Facteurs Affectant Le Pronostic
Bien que les RMS soient regroupés en une seule maladie, il existe des différences importantes dans le comportement de ces tumeurs qui dépendent de leur emplacement, leurs caractéristiques microscopiques, leur taille, l'absence ou présence de métastases, le nombre de cellules cancéreuses encore présentes suite à la chirurgie initiale, l'âge du patient au moment du diagnostic. Ces facteurs sont dits "pronostiques". Ils permettent d'évaluer les "probabilités statistiques" de guérison. Cependant, quelle que soit l'influence de ces facteurs sur le pronostic, ils ne peuvent en aucun cas déterminer la guérison d'un individu en particulier.
Le tableau 2 résume comment l'association des facteurs : emplacement, taille de la tumeur, état régional des ganglions, présence de métastases distantes, âge du patient lors du diagnostic et histologie, sont utilisés pour décider quel est le traitement approprié par rapport au risque pour les patients atteints de RMS. La colonne "risque" sépare les patients en quatre groupes de niveau de risques différents (Peu élevé-A, Peu élevé-B, Intermédiaire, Elevé) qui sont utilisés pour attribuer le traitement adéquat lors de l'étude "Fifth Intergroup Rhabdomyosarcoma Study" (IRS-V). Le numéro spécifique du protocole est indiqué entre parenthèses avec la lettre D suivie de 4 chiffres.
D9602 représente l'étude des groupes à risque peu élevé qui consiste à environ 11 mois de chimiothérapie de 2 régimes différents, soit le régime A (vincristine + dactinomycin [VA], avec ou sans radiothérapie) soit le régime B (vincristine + dactinomycin + cyclophosphamide [VAC], avec radiothérapie pour presque tous les patients). D9803représente l'étude du groupe à "risque intermédiaire" qui consiste à randomiser l'attribution de 2 types de chimiothérapie dont l'une est le régime A (14 cycles de VAC) et l'autre le régime B (8 cycles de VAC alternés avec 6 cycles de vincristine + topotecan + cyclophosphamide). Ce groupe reçoit également de la radiothérapie. D9802 est l'étude du groupe à "risque élevé" qui consiste à "une fenêtre de phase II" de l'irinotecan administré sur un "programme journalier x5x2", développé dans le laboratoire Houghton du centre de recherche St. Jude Children’s Research Center (réf. 13), soit tout seul, soit en combinaison avec la vincristine, suivi de, soit 8 cycles de VAC plus 4 cycles de vincristine plus Irinotecan pour les patients qui répondent à l'Irinotecan, soit 12 cycles de VAC pour les patients qui ne répondent pas à l'Irinotecan, plus de la radiothérapie. Les différentes études IRS-V sont supposées finir le recrutement de patients avant la fin 2004. Les études successives doivent débuter en 2005-2006.
Définitions des Groupes à Risques des RMS
Favorable = Orbite/paupière, tête et cou (à l'exclusion des localisations paraméningées), génito-urinaires (mis à part la vessie et la prostate)
Défavorable = Vessie, prostate, membres, paraméninges, autres (tronc, rétropéritoine, etc...)
a = taille de la tumeur ≤ 5 cm de diamètre
b = taille de la tumeur > 5 cm de diamètre
EMB = embryonnaires, botryoïdes ou variantes fusiformes ou ectomésenchymes avec caractéristiques embryonnaires
ALV = sarcomes alvéolaires et indifférenciés ou ectomésenchymes avec caractéristiques alvéolaires
N0 = Aucune atteinte métastatique des ganglions régionaux
N1 = Envahissement ganglionnaire régional
NX = Statut des ganglions non connu
| Risque | Stade | Groupe | Emplacement | Taille | Age | Histologie | Métastases | Ganglions |
|---|---|---|---|---|---|---|---|---|
| Peu élevé A (D9602) | 1 | I | favorable | a ou b |
<21 | EMB | M0 | N0 ou N1 ou NX |
| 1 | II | favorable | a ou b | <21 | EMB | M0 | N0 ou NX | |
| 1 | III | seulement l'orbite | a ou b | <21 | EMB | M0 | N0 ou NX | |
| 2 | I | défavorable | a | <21 | EMB | M0 | N0 ou NX | |
| Peu élevé B (D9602) | 1 | II | favorable | a or b | <21 | EMB | M0 | N1 |
| 1 | III | seulement l'orbite | a or b | <21 | EMB | M0 | N1 | |
| 1 | III | favorable (exclut l'orbite) | a or b | <21 | EMB | M0 | N0 ou N1 ou NX | |
| 2 | II | défavorable | a | <21 | EMB | M0 | N0 ou NX | |
| 3 | I or II | défavorable | a | <21 | EMB | M0 | N1 | |
| 3 | I or II | défavorable | a | <21 | EMB | M0 | N0 ou N1 ou NX | |
| Intermédiaire (D9803) | 2 | III | défavorable | a | <21 | EMB | M0 | N0 ou NX |
| 3 | III | défavorable | a | <21 | EMB | M0 | N1 | |
| 3 | III | défavorable | b | <21 | EMB | M0 | N0 ou N1 ou NX | |
| 1 or 2 or 3 | I or II or III | favorable ou défavorable | a ou b | <21 | ALV | M0 | N0 ou N1 ou NX | |
| 4 | IV | favorable ou défavorable | a ou b | <10 | EMB | M1 | N0 ou N1 ou NX | |
| Elevé (D9802) | 4 | IV | favorable ou défavorable | a ou b | >=10 | EMB | M1 | N0 ou N1 ou NX |
| 4 | IV | favorable ou défavorable | a ou b | <21 | ALV | M1 | N0 ou N1 ou NX |
Les cancérologues utilisent une série d'abréviations spéciales pour décrire ces facteurs. Pour les enfants atteints de RMS, il existe 2 types de terminologie pour décrire ces facteurs. L'une est appelée stade et l'autre groupe clinique (ou "groupe" pour abréger). Le stade dépend de 3 facteurs:
- La partie du corps dans laquelle la tumeur initiale est apparue.
- La taille de la tumeur initiale.
- La présence/absence de métastases locales ou distantes (voir tableau 3).
Le groupe dépend de la quantité de tumeur restante après la chirurgie initiale. Il existe 4 stades (stades 1, 2, 3 et 4) et 4 groupes (groupes I, II, III et IV). Chaque patient se voit attribué un stade et un groupe après identification de ces facteurs.
Les tableaux 3 et 4 contiennent les détails du système de classification du stade et du groupe utilisés pour les patients atteints de RMS. Ce système "simplifié" est un des aspects les plus déboussolant du soin des enfants atteints de RMS. Toute tumeur se présentant dans un des emplacements favorables est considérée de stade 1, du moment qu'il n'y a pas de métastase distante. Toute tumeur qui s'est propagée dans une partie "distante" du corps, est considérée de stade 4 . Les tumeurs qui se développent dans un des emplacements défavorables sont classées en stade 2 (si elles sont "petites" et n'ont pas atteint les ganglions) ou stade 3 (si elles sont "grosses" ou ont atteint les ganglions). La plupart des enfants atteints de RMS ont des tumeurs de stade 2 ou stade 3. Puisque la classification du stade n'exige pas une confirmation pathologique des anomalies identifiées par imagerie, des problèmes de pertinence de la classification peuvent se présenter lorsque par exemple, un patient est classé en stade 4 de par la présence d'un nodule pulmonaire visible au scanner qui est interprété comme étant une métastase mais qui, à la suite de l’intervention chirurgicale, s'avère bénin.
| Stade | Emplacement | Statut T | Taille | Etat des ganglions | Métastases |
|---|---|---|---|---|---|
| 1 | Favorable | T1 ou T2 | a ou b | N0, N1, ou NX | M0 |
| 2 | Défavorable | T1 ou T2 | a | N0 ou NX | M0 |
| 3 | Défavorable | T1 ou T2 | a | N1 | M0 |
| 3 | Défavorable | T1 ou T2 | b | N0, N1, ou NX | M0 |
| 4 | Favorable ou Défavorable | T1 ou T2 | a ou b | N0 ou N1 | M1 |
Toute tumeur qui a été enlevée dans sa totalité au moment de chirurgie initiale est considérée de groupe I. Une tumeur qui a visiblement atteint une autre partie "distante" du corps est toujours considérée de groupe IV. Une tumeur qui est encore visible après la chirurgie d’exérèse (sur le scanner ou lors de l'examen physique) est considérée de groupe III. Le groupe II est utilisé lorsque toute la tumeur visible a été enlevée mais qu’il reste des cellules tumorales en quantité "microscopique" avec ou sans atteinte des ganglions régionaux (du moment qu'ils ont été enlevés). La moitié des enfants atteints de RMS ont des tumeurs de groupe III.
| Groupe clinique | Définition |
|---|---|
| I | Résection complète, marges négatives |
| IIa | Résection complète, marges négatives |
| IIb | Résection complète, marges négatives, ganglions réséqués positifs |
| IIc | Résection complète, marges négatives, ganglions réséqués positifs |
| III | Reste de tumeur visible (inclut les ganglions régionaux non réséqués) |
| IV | Métastases distantes |
Modes de Propagation
Les RMS peuvent se propager localement, de façon régionale ou à distance.
- Une propagation locale signifie que la tumeur infiltre ou envahit le tissu à proximité immédiate de l'emplacement initial.
- Une propagation régionale signifie que la tumeur a atteint les ganglions qui drainent la zone dans laquelle elle est apparue. Une telle propagation est le plus probable chez les enfants atteints de tumeurs aux membres et chez les garçons plus âgés (10 ans ou plus) atteints de tumeurs para-testiculaires.
- Une propagation à distance signifie que la tumeur a voyagé dans les vaisseaux sanguins et atteint une partie différente du corps. Dans ce cas, les destinations les plus probables sont les poumons, les os et la moelle osseuse.
Les RMS se propagent rarement au cerveau ou aux autres organes tels que le foie ou la rate. Les tumeurs qui ont visiblement atteint des localisations distantes sont appelées "métastases". Seulement un enfant sur cinq développe des métastases distantes.
Une gamme de tests est nécessaire pour analyser la tumeur d'origine et la présence de métastases. La première évaluation passe toujours par un historique complet et un examen physique. Généralement, la meilleure technique d’imagerie pour l'évaluation de la tumeur initiale est l'IRM. Elle fournit une image en 3 dimensions et est fréquemment utile pour de la préparation de la radiothérapie ou de la chirurgie. Les scanners du thorax sont régulièrement utilisés pour vérifier si des métastases ont atteint les poumons. Suivant l'emplacement de la tumeur d'origine, des scanners de l'abdomen et du pelvis sont parfois utilisés pour vérifier si les ganglions sont atteints. Une scintigraphie osseuse est une technique d’imagerie de médecine nucléaire qui permet d’étudier la totalité du squelette pour voir si la tumeur s'est propagée aux os. Un autre test de médecine nucléaire qui est de plus en plus utilisé, est la tomographie à émission de positon (TEP ou PET). Ce test est relativement unique car il donne une image du corps complet montrant les os et les tissus mous et permet de clarifier les résultats ambigus d'un scanner ou d’une IRM et peut également aider à évaluer la réponse à un traitement. Etant donné que les RMS peuvent se propager à la moelle osseuse, des ponctions et des biopsies de moelle osseuse sont également faites chez les patients atteints de RMS. Une aiguille est placée au niveau des os de la hanche et un échantillon de moelle est prélevé pour être testé. Ces tests sont toujours réalisés au moment où l'anesthésie est administrée pour la biopsie de la tumeur ou pour l'insertion du cathéter veineux central (CVC). Les patients dont les tumeurs se développent dans une des localisations paraméningées doivent toujours subir une ponction lombaire pour l'obtention d'un échantillon du liquide cérébro-spinal (LCR) dans le but de vérifier si la tumeur a infiltré l'enveloppe du cerveau.
Environ 20% des nouveaux patients se présentent avec un ou plusieurs sites de métastases distantes. Un nombre disproportionné de ces patients sont atteints de tumeurs alvéolaires. Des 127 patients atteints de RMS métastatique traités lors de l'étude IRS-IV, 46% des patients présentaient des tumeurs alvéolaires alors que ceux-ci ne représentaient que 22% des 900 patients dépourvus de métastases. (réf. 14). Près de 60% des patients avaient des métastases limitées à un seul emplacement. Le site le plus commun était les poumons (39%), suivis de la moelle osseuse (32%), des ganglions (30%) et des os (27%). Bien que les ponctions et biopsies de moelle osseuse soient régulièrement recommandées pour la classification des nouveaux patients atteints de RMS, connu ou suspecté, une atteinte isolée de la moelle osseuse ne s'est présenté que chez 12 des 900 patients sans autre site de métastase connu. Le rendement de la ponction et de la biopsie de la moelle osseuse pour les patients atteints de RMS localisé est donc de moins de 2%.
Principes Generaux Pour Le Traitement Des Rms
Tous les enfants atteints de RMS sont traités par chimiothérapie. Suivant la taille et l'emplacement de la tumeur initiale et à quel point elle peut être réséquée par chirurgie, la plupart des enfants subissent également une combinaison de radiothérapie et de chirurgie.
Chirurgie
Le diagnostic d'un RMS ne peut se faire sans passer par l'étude d'un échantillon de la tumeur en laboratoire. Le procédé initial pour obtenir un échantillon est appelé biopsie. Une biopsie est généralement considérée comme une "petite" opération. La plupart du temps, elle ne nécessite pas de passer la nuit à l'hôpital. Il existe différents types de biopsies:
- La biopsie percutanée à l'aiguille: au cours de cette procédure, une aiguille est placée dans la tumeur au travers de la peau et un petit morceau de tumeur est prélevé à l'intérieur de l'aiguille. Parfois, cette procédure est guidée par échographie ou scanner (tomodensitomètre). Cette procédure ne nécessite généralement pas d'anesthésie bien qu'une sédation intraveineuse puisse être requise suivant la localisation de la tumeur et l'âge de l'enfant. Suivant l'emplacement de la tumeur, cette procédure peut être plus ou moins sûre que les autres types de procédures décrites ultérieurement. Une biopsie à aiguille est capable de fournir un échantillon adéquat dans 90% des cas.
- La biopsie chirurgicale incisionnelle: au cours de cette procédure qui est pratiquement toujours entreprise sous anesthésie, une petite incision est faite dans la peau par laquelle un petit morceau de tumeur est enlevé. Cette procédure fournit un échantillon adéquat pour l'obtention d'un diagnostic correct dans 100% des cas.
- La biopsie chirurgicale excisionnelle: au cours de cette procédure qui est toujours entreprise sous anesthésie, une incision est faite dans la peau pour tenter l'extraction de la tumeur dans sa totalité. Cette opération est plus importante que les deux précédentes. Elle est adéquate pour les enfants dont les tumeurs ont été pleinement étudiées par imagerie médicale et le chirurgien est sûr que la tumeur peut être enlevée dans sa totalité sans produire un déficit fonctionnel (autrement dit, si une tumeur du mollet peut être réséquée sans amputation et sans compromettre la capacité de se déplacer) ou un défaut cosmétique (autrement dit, si une tumeur des sinus peut être extraite sans produire une grosse cicatrice ou déformation du visage).
Puisque l'imagerie peut souvent ne pas détecter la présence de tumeur dans les ganglions régionaux, leur évaluation chirurgicale est obligatoire dans 2 cas spécifiques, celui des enfants atteints de RMS au niveau des membres et celui des garçons de 10 ans ou plus atteints de tumeurs paratesticulaires. Pour les RMS des membres, un prélèvement chirurgical des ganglions situés derrière le genou ou à l'aine est effectué pour les tumeurs des membres inférieurs et un prélèvement derrière le coude ou aux aisselles est effectué pour les tumeurs des membres supérieurs (réf. 15). Le rôle de la lymphoscintigraphie pour identifier un ganglion sentinelle est en train d'être développé. La tomographie à émission de positon peut être utile pour l'identification de ganglions inquiétants qui autrement n'apparaissent pas clairement sur les images conventionnelles des scanners ou IRM.
Dans le cas des garçons atteints de tumeurs paratesticulaires, le prélèvement des ganglions doit idéalement être entrepris au même moment que la résection de la tumeur d'origine. Une incision inguinale est effectuée comme pour l'opération d'une hernie et la tumeur et les testicules doivent être extraits et réséqués ensemble de l'intérieur du scrotum. Le prélèvement des ganglions ipsilatéraux (du même côté que la tumeur) du rétropéritoine doit être effectué. Cette procédure est de plus en plus souvent réalisée par laparoscopie pour minimiser le temps de rétablissement après l'opération et potentiellement rapprocher le moment de l'administration de la chimiothérapie (réf. 16).
Il est important de rappeler que la chirurgie n'est jamais curative pour les enfants atteints de RMS. Il est aussi important de rappeler que le rôle de la chirurgie dépend de l'emplacement de la tumeur. Alors qu' une résection complète d'une tumeur des membres ou du pelvis améliore les chances de guérison, la résection complète d'une tumeur orbitale ou du vagin n'est presque jamais nécessaire pour atteindre un taux très élevé de guérison (et n'est presque jamais adéquate). Bien que la plupart des familles dont l'enfant est suspecté d'être atteint d'un RMS, veulent que la tumeur soit enlevée dans sa totalité le plus rapidement possible, la chirurgie initiale n'est presque jamais une urgence et il est impératif d'obtenir des images adéquates de la tumeur avant la réalisation d'une biopsie si un RMS est suspecté. L'obtention d'images de la tumeur initiale avant la biopsie est nécessaire pour la planification adéquate d'une radiothérapie dont la nécessité peut être critique. De façon similaire, il est important que la biopsie soit obtenue dans un centre où les anatomo-pathologistes sont expérimentés et sont capables de procéder rapidement et minutieusement à l'examen complet de l'échantillon (réf. 17).
Anatomo-Pathologie des RMS

Après une biopsie, la tumeur est étudiée sous le microscope dans un laboratoire. Les caractéristiques qui permettent de conclure à un diagnostique de RMS doivent porter la preuve que la tumeur appartient à la lignée du muscle squelettique, soit par son apparence sous le microscope, soit par sa réponse à l'immunomarquage. Il existe deux catégories de RMS : embryonnaire et alvéolaire. Environ 2/3 des enfants souffrant d’un RMS sont atteints du type embryonnaire (ou de la variante à cellule fusiforme ou botryoïde). Ces tumeurs sont plus fréquentes chez les enfants plus jeunes, en particulier lorsque les tumeurs se développent au niveau de la tête ou du cou (ce qui inclut les localisations paraméningées) et au niveau du système génito-urinaire (ce qui inclut la vessie et la prostate). Les cellules ont tendance à être plus longues et se présentent en moindre densité.

Environ 20 à 25 % des enfants atteints de RMS présentent le type alvéolaire (ou la variante alvéolaire de forme solide). Ces tumeurs sont beaucoup plus communes chez les adolescents et se présentent le plus souvent au niveau des membres. Les cellules tumorales ont tendance à être plus petites et rondes, souvent avec une densité cellulaire plus importante, et sont ainsi nommées à cause de leur ressemblance avec les petits sacs d'air des poumons (les "alvéoles"). Les tumeurs alvéolaires sont souvent considérées plus "agressives" ou à "risque plus élevé" que les tumeurs embryonnaires, en particulier pour les tumeurs qui se présentent dans une des localisations favorables.
Environ 5 à 10% des enfants ont des tumeurs qui ne peuvent pas être placées dans une de ces catégories et sont considérées soit "indifférenciées" ou "rhabdomyosarcome, non spécifié."
Lorsqu'un échantillon d'une tumeur a été prélevé par biopsie et l'anatomo-pathologiste (le médecin qui étudie la tumeur dans le laboratoire) soupçonne un RMS, celui-ci procède généralement à des tests appelés «immunomarquages» pour confirmer le diagnostic . Ceux-ci emploient des réactions chimiques qui "marquent" les différentes structures des cellules tumorales. Les tumeurs des RMS sont généralement positives à plusieurs immunomarquages dont la desmine et la myogénine. Un marquage positif à la myogénine permet virtuellement le diagnostic d'un RMS.

Une dernière série de tests est parfois effectuée sur les cellules tumorales des RMS. Il s'agit des tests de diagnostic moléculaire. Bien que l'on ne sache pas trop pourquoi une cellule du muscle squelettique normale devient cancéreuse, les changements génétiques qui se produisent dans la cellule une fois qu'elle devient cancéreuse sont plutôt bien connus. Dans pratiquement tous les cas de RMS embryonnaires, une anomalie peut être détectée dans les cellules cancéreuses (et seulement dans les cellules cancéreuses ce qui veut dire que cette anomalie n'est pas héritée génétiquement!). Cette anomalie provoque un sur-expression d'un gène qui est important pour la croissance des cellules du muscle normal.
Les RMS embryonnaires se présentent généralement avec une sur-expression du gène de l'IGF-2 situé sur le bras court du chromosome 11. Ceci serait le résultat de la perte de l'allèle maternel et duplication de l'allèle paternel. L'expression des deux copies du gène semble mener à un effet de surdosage où trop de IGF-2 produit un constant signal de prolifération qui permet aux cellules musculaires pré-cancéreuses (ou déjà cancéreuses) de se développer sans contrainte et les protège dans un environnement de stress qui devrait leur être normalement fatal. Ce processus est connu sous le nom de "perte d'hétérozygotie".

Ce processus entraîne un "surdosage" d'un "gène promoteur de croissance", le facteur de croissance analogue à l'insuline de type 2 (IGF-2) qui se trouve sur le chromosome 11. Normalement, une seule copie (généralement celle héritée du père) du gène est "active" et l'autre est "silencieuse" (une modification chimique de la structure de l'ADN proche du gène, connue sous le nom de " méthylation" semble être responsable de l'activation d'un des gènes et l'inactivation du gène voisin, suppresseur de croissance [H19]). Dans la plupart des cas de RMS embryonnaires, soit les 2 gènes sont activés ou la copie du gène maternel est perdue alors que le gène paternel est dupliqué et ses 2 copies sont "actives". Cela semble mener à la production d'un signal constant de "prolifération" qui signale à la cellule de continuer de croître et la protège des pressions environnementales normales qu'elle subit.
De nombreux cancers pédiatriques sont associés à des translocations spécifiques où un morceau d'un gène normal et un morceau d'un autre gène normal se brisent et échangent leur place. Près de 90% des cas de RMS alvéolaires, une portion de l'un des gènes PAX (le plus souvent le gène PAX 3 situé sur le chromosome 2, ou moins souvent le gène PAX 7 situé sur le chromosome 1) fusionne avec une portion du gène FKHR (situé sur le chromosome 13) pour former un nouveau gène "hybride" (PAX-FKHR) qui active des gènes stimulateurs de croissance qui sont normalement "inactifs" et désactive des gènes inhibiteurs de croissance qui sont normalement actifs. Ce gène "hybride" n'est présent que dans les cas de RMS alvéolaires et peut donc être utilisé pour le diagnostic et devenir dans le futur une cible pour les thérapies utilisant le système immunitaire.
Cette anomalie peut souvent être détectée en utilisant l'une des diverses techniques spécialisées qui examinent le contenu des chromosomes des cellules tumorales.
Une translocation est un phénomène relativement courant des cancers de l'enfant où un morceau d'un gène normal se détache de son emplacement habituel et se fixe à un morceau d'un autre gène normal. Plus précisément, en fusionnant les domaines "paired-box" (PB) et "homéobox" (HD) du gène Pax 3 avec le domaine d'activation transcriptionnelle (TAD) du gène FKHR, un nouveau gène "hybride" est créé qui semble jouer de deux manières un rôle crucial dans le processus par lequel la cellule d'un RMS devient cancéreuse. Tout d'abord, il inhibe d'autres gènes qui sont normalement "actifs" et servent de «freins» à la croissance cellulaire. Deuxièmement, il active d'autres gènes qui sont normalement "inhibés" et servent de stimulateurs de croissance, survie et propagation cellulaires. Cette anomalie n'est jamais présente chez les RMS embryonnaires et donc lorsque l'apparence sous le microscope laisse un doute, la démonstration de la présence de la translocation PAX-FKHR permet de conclure à un RMS alvéolaire. Cette anomalie est généralement testée par l'utilisation d'une technique appelée RT-PCR (reverse transcriptase polymerase chain reaction). Cependant, cet test peut n'être disponible que dans les laboratoires spécialisés de grands centres cancéreux ou hôpitaux pour enfants.
Les deux sous-types histologiques principaux des RMS, à savoir embryonnaire et alvéolaire, présentent des altérations génétiques spécifiques mais distinctes qui sont censées jouer un rôle dans la pathogenèse de ces tumeurs. Le RMS alvéolaire possède une translocation spécifique entre le bras long du chromosome 2 et le bras long du chromosome 13, identifiée par la notation t(2; 13) (q35; q14), voir réf. 18-19. Le clonage moléculaire de cette translocation a démontré l'implication de la juxtaposition du gène PAX3 (ou, rarement, le gène Pax7 situé sur le chromosome 1p36), estimé réguler la transcription au cours du développement neuro-musculaire initial, et le gène FKHR, également connu sous le nom FOXO1a , un membre de la famille des facteurs de transcription "forkhead" (réf. 20-21). Ce facteur de transcription issu d'une fusion serait responsable de l'activation anormale de la transcription d'un gène ou de gènes qui contribueraient au phénotype transformé. Bien que la conséquence exacte de cette translocation spécifique de la tumeur reste à élucider, il a été démontré en utilisant l'analyse des puces à ADN que la fusion PAX-FKHR, exprimée dans le cas des fibroblastes, active un ensemble de facteurs myogéniques (réf. 22). De plus, PAX-3-FKHR est responsable d'une augmentation de l'expression de c-MET, un récepteur tyrosine kinase qui est impliqué dans la transformation (réf. 23). L'utilisation de la réaction en chaîne par polymérase (PCR) pour la confirmation précise du diagnostic génétique d'un RMS alvéolaire devrait se répandre dans un proche avenir. Récemment, un nouvel amplicon a été identifié au niveau de 13q31 dans environ 20% des cas de RMS alvéolaires, ce qui suggère que un ou plusieurs gènes de ce locus contribuent à la pathogenèse de ces tumeurs (réf. 24).
L'autre important sous-type histologique, le RMS embryonnaire, présente une perte d'hétérozygotie (LOH) au locus 11p15 (réf. 25-26). En outre, il a été démontré que cette LOH se traduit par la perte de l'information génétique de la mère et la duplication du matériel génétique paternel à ce locus (réf. 27). Cette région est particulièrement intéressante parce que c'est la localisation du gène IGF-2, qui code pour un facteur de croissance qui semble jouer un rôle dans la pathogenèse des RMS (voir plus loin). IGF-2 est un gène soumis à empreinte avec seulement l'allèle paternel transcriptionnellement actif (réf. 28-29). Il est donc concevable que, pour cette tumeur, LOH avec disomie paternelle peut conduire à la surexpression de IGF-2. Toutefois, il est également possible que LOH au locus 11p15 reflète la perte d'une activité de suppresseur de tumeur qui n'a pas été identifiée, ou que l'activation de IGF-2 et la perte de suppresseur de tumeur résultent toutes les deux de LOH au locus 11p15 pour les RMS embryonnaires (réf. 30).
Plusieurs chercheurs ont récemment rapporté des résultats obtenus par hybridation génomique comparative (CGH) des tumeurs et cellules RMS. Trois caractéristiques ont été ainsi mises en évidence. Premièrement, des régions d'amplifications génomiques sont observées dans les RMS alvéolaires et les RMS embryonnaires anaplasiques, suggérant que ces sous-types partagent les mêmes événements génétiques (réf. 31). Deuxièmement, plusieurs études ont noté l'amplification significative de 15q25-26, le locus du récepteur de l'IGF-1 (réf. 24, 31) et une amplification spécifique de IGF-1R a été confirmée par PCR et FISH (réf. 31). Il est important de le noter puisque la signalisation de IGF est impliquée dans les RMS. Enfin, deux études ont démontré la perte du locus 9q22 dans environ 33% des tumeurs. Cette région correspond au locus PTH (hormone parathyroïde), un gène suppresseur de tumeur impliqué dans le développement des RMS dans un modèle murin du syndrome de Gorlin (réf. 31-32).
Une fois que toutes les études d'imagerie ont été achevées, la biopsie effectuée et le diagnostic de RMS confirmé, il est possible de classer les patients atteints de RMS dans l'un des quatre "groupes à risque" en se basant sur la combinaison de leur stade (localisation, taille, envahissement ganglionnaire), leur groupe (mesure de la tumeur résiduelle post-opératoire), leur âge au moment du diagnostic, leur sous-type histologique (embryonnaire versus alvéolaire) et la présence ou absence de métastases à distance. Ces groupes de risque fournissent des informations importantes sur la curabilité potentielle de la tumeur selon des traitements d'intensité plus ou moins grande:
- Risque standard , sous-groupe A: ces patients, relativement peu nombreux, ont un taux survie de plus de 85% après un traitement relativement peu intensif comportant une chimiothérapie utilisant 2 médicaments, avec ou sans radiothérapie. Ce groupe est essentiellement composé de patients atteints de tumeurs orbitaires (du moment qu'elles n'ont pas métastasé), de patients aux tumeurs de localisation "favorable" (stade 1) qui ont été soit complètement enlevées par chirurgie(Groupe I) ou seulement un résidu microscopique de la maladie est encore présent (Groupe II) et de patients dont les petites tumeurs de localisation défavorable (stade 2) ont été complètement réséquées (groupe I).
- Risque standard , sous-groupe B: ces patients, un peu plus nombreux, ont un taux de survie de plus de 80%, mais un besoin relativement plus important, une chimiothérapie à 3 médicaments, généralement avec de la radiothérapie (avec une exception importante, voir ci-dessous). Ce groupe est composé de tous les patients avec des tumeurs non métastatiques, non-orbitaire, de localisation favorable (stade 1) qui sont encore visibles (Groupe III) après la chirurgie initiale, des patients avec des petites tumeurs non métastatiques de localisation défavorable sans propagation ganglionnaire régionale (stade 2) dont la partie visible a été totalement réséquée (Groupe 2) et des patients atteints de tumeurs de localisation défavorable qui sont de taille importante ou se sont propagées aux ganglions régionaux (stade 3) mais ont été complètement ou en apparence totalement réséquées (Groupes I et II). Environ 15 à 20% des patients nouvellement diagnostiqués d'un RMS seront considérés comme de risque standard. Les patients atteints de RMS alvéolaires ne sont jamais considérés comme de risque standard.
- Risque intermédiaire: Ces patients représentent la majorité des patients atteints de RMS nouvellement diagnostiqués et comprennent ceux qui sont atteints de tumeurs aux localisations défavorables (stades 2 et 3) qui n'ont pas été complètement réséquées (Groupe III), des patients de moins de dix ans atteints de RMS embryonnaire qui s'est propagé à d'autres parties du corps (stade 4, groupe IV) et de tous les patients atteints de RMS alvéolaire non métastatique. Bien que ce groupe de patients soit diversifié, le pronostic de guérison avec une chimiothérapie à 3 médicaments (ou plus) et une radiothérapie dépasse généralement les 50% et peut même atteindre 70% pour certains sous-groupes.
- Risque élevé: ces patients représentent environ 10 à 15% des patients nouvellement diagnostiqués avec un RMS. Le pronostic de guérison de ces enfants est généralement assez faible, entre 20% et 35%, même avec une chimiothérapie très agressive, de la radiothérapie et une chirurgie. Ce groupe comprend tous les patients atteints de RMS alvéolaire métastastique, les patients âgés de dix ans ou plus atteints de RMS embryonnaire métastatique et probablement deux autres groupes actuellement considérés comme à risque intermédiaire: les nourrissons de moins d'un an atteints de RMS embryonnaire métastatique, dont le taux de survie à 5 ans est inférieur à 20% (réf. 33) et les enfants atteints de tumeurs aux extrémités avec une propagation ganglionnaire régionale, la quasi-totalité d'entre eux atteints d'un RMS alvéolaire, pour qui le taux de survie à 5 ans est d'environ 30% (réf. 15).
Traitement des Rhabdomyosarcomes
Le traitement des patients atteints de RMS est pluridisciplinaire et commence même avant le début du traitement avec l’implication de radiologues qualifiés qui peuvent interpréter correctement les résultats d'imagerie, d'anatomo-pathologistes qualifiés qui sont familiers avec l'évaluation et les tests des tumeurs pédiatriques à petites cellules rondes bleues et des chirurgiens qualifiés qui comprennent le rôle de la chirurgie initiale dans la gestion globale des patients atteints de RMS. Il comprend les radiothérapeutes-oncologues et les cancérologues pédiatriques qui sont familiers avec les directives nationales (ou institutionnelles) de traitement (également connues sous le nom de standards) pour le traitement de cette forme rare de cancer. Idéalement, le traitement sera donné dans un centre où des réunions régulières de l'ensemble de ces disciplines (connues sous le nom de réunion de concertation pluridisciplinaire=RCP) sont organisées pour que tous les professionnels de santé impliqués dans les soins de l'enfant puissent voir les résultats les images, les biopsies et les évaluations pendant le traitement qui sont nécessaires pour fournir des soins optimaux.
Étant donné le jeune âge de ces patients, l'équipe de traitement devrait également intégrer des anesthésiologistes pour maintenir les patients calmes lors des examens et procédures (y compris parfois pour l'ensemble des 5 à 6 semaines de radiothérapie) et un personnel infirmier familiarisé avec les besoins et complications médicaux particuliers aux enfants atteints de cancer. Enfin, elle comprend un personnel d'assistance sociale, de soutien psychologique et éducatif à l'enfant et des aumôniers pour aider la famille (et l'enfant) dont le monde a été brisé par les mots "votre enfant a un cancer."
Le but du traitement des enfants atteints de RMS est de parvenir à un "contrôle local" et un "contrôle systémique". Un "contrôle local" fait référence à l'élimination permanente de la "tumeur primaire". Ceci est habituellement réalisé par l'ablation chirurgicale ou l'irradiation (ou les deux) de la tumeur et des zones voisines concernées, en plus de la chimiothérapie. Un "contrôle systémique" fait référence généralement au contrôle permanent des "micrométastases" invisibles ou "métastases" visible, par chimiothérapie (avec parfois une intervention chirurgicale ou une radiothérapie). Le risque que le traitement ne puisse pas être curatif varie en fonction du "groupe de risque". Pour la plupart des enfants atteints de tumeurs non métastatiques (c'est-à-dire à risques standard et intermédiaire), le plus grand risque est que la tumeur primaire ne soit pas contrôlée du premier coup. Plus de la moitié de des échecs de traitement dans ces groupes sont "loco-régionaux" (c'est-à-dire, à ou près de l'emplacement d'origine). L'incapacité de contrôler la tumeur initiale est associée à une augmentation marquée du risque de rechute dans d'autres parties du corps ce qui est probablement un reflet d'une résistance à la chimiothérapie et la radiothérapie, intrinsèque ou rapidement acquise. Pour la plupart des enfants atteints de tumeurs métastatiques (c'est-à-dire à risque élevé), l'échec du traitement est généralement dû au fait que les métastases ne sont pas contrôlées même si la tumeur d'origine est contrôlée. Bien qu'il existe des exceptions, parce que la survie suite à une rechute est faible pour la grande majorité des enfants atteints de RMS récurrents (moins de 20% des patients qui rechutent seront guéris), il est essentiel qu'un traitement optimal soit fournit au moment du diagnostic.
Traitement
Le traitement de la plupart des enfants atteints de RMS est administré soit par le biais d'un essai clinique d'un groupe d'établissements en coopération, d'un établissement individuel ou un groupe limité d'établissements ou en suivant les directives de l'essai approprié. Depuis 1972, "l'Intergroupe Rhabdomyosarcome Study Group" (IRSG) a réalisé quatre grands essais cliniques séquentiels et prospectifs traitant plus de 4000 patients atteints de RMS. Pour les patients présentant des tumeurs non métastatiques, l'étude la plus récemment achevée, IRS-IV, cherchait à répondre à deux grands questions par le biais de la recherche (aléatoire):
- Le remplacement de la cyclophosphamide par l'ifosfamide (VAI), ou de la dactinomycine par l'étoposide (VIE) peut-il améliorer les résultats pour les enfants atteints de tumeurs du groupe III par rapport à ceux qui reçoivent une chimiothérapie standard (VAC)?
- La radiothérapie hyperfractionnée (5940 cGy administrés en fractions de 110 cGy, deux fois par jour) peut-elle améliorer le contrôle local par rapport aux radiations conventionnelles (5040 cGy administrés en fractions quotidiennes de 180 cGy)?
Pour les patients atteints de tumeurs métastatiques, l'essai clinique le plus récemment achevé tentait d'évaluer l'activité anti-tumorale et l'efficacité finale du traitement par l'une des trois paires de deux médicaments (ifosfamide plus doxorubicine, vincristine plus melphalan ou ifosfamide plus étoposide) administrée en plus de la chimiothérapie "classique" VAC.
Les résultats de ces études ont été publiés au cours des dernières années (réf. 14 et réf. 34-38). Pour les enfants atteints de tumeurs non métastatiques, aucune différence dans les résultats n'a été observée entre les trois types de chimiothérapies: VIE, VAI, VAC (réf. 35). Sur cette base, la chimiothérapie VAC a continué à être recommandée par le IRSG comme le "meilleur standard" pour les enfants atteints de RMS. Comparé à l'étude précédente, IRS-III, le résultat a été amélioré pour seulement un petit nombre d'enfants atteints de tumeurs embryonnaires, ceux aux tumeurs non réséquées se présentant dans des localisations "favorables" (Groupe III) et ceux aux tumeurs de localisations "défavorables" (stades 2 et 3) complètement ou visuellement totalement réséquées (Groupes I et II) (réf. 34). La survie sans échec à 3 ans [SSE] pour tout le groupe de patients était de 77%. Les résultats des patients à l'histologie alvéolaire étaient nettement plus mauvais (66% SSE à 3 ans contre 83% pour les patients atteints de tumeurs embryonnaires). La radiothérapie hyperfractionnée n'a pas produit des taux supérieurs de contrôle local (et n'a pas eu un impact sur la survie globale) par rapport à la radiothérapie conventionnelle fractionnée (réf. 36). Le taux global de contrôle local était de 87%. Le plus grand risque d'échec du traitement local (récidive locale) a été observé chez les patients aux tumeurs de la vessie/prostate (19%) et tumeurs paraméningées (16%).
Pour les patients avec des métastases, bien que les combinaisons de médicaments aient été toutes les trois très actives, avec des taux de réponse de 60 à 80% (réf. 37-38), les évolutions sont restées mauvaises. La survie globale pour l'ensemble du groupe était inférieure à 30%. De meilleurs résultats ont été suggérés chez les patients recevant IE, en plus de VAC (réf. 38). L'utilisation de melphalan a été trouvée associée à une déficience de la tolérance des chimiothérapies subséquentes. Bien que les résultats pour les patients atteints de RMS métastatique restent mauvais, aucun avantage n'a été trouvé à consolider le traitement avec une chimiothérapie à haute dose et une autogreffe de moëlle osseuse en support (réf. 39).
Chimiotherapie
Aux États-Unis, la plupart des enfants sont traités suivant le protocole d'un essai clinique international précédemment connu sous le nom de "Intergroup Rhabdomyosarcoma Study" (maintenant connu sous le nom de "Soft Tissue Sarcoma Committee of the Children’s Oncology Group"). Au cours des 30 dernières années, quatre études de ce groupe ont été finalisées traitant plus de 4000 patients atteints de RMS. La 5ème génération de ces études devrait achever son recrutement cette année. Pour certains patients, généralement ceux atteints de RMS à risque intermédiaire ou élevé, un traitement en essai clinique dans un établissement «pilote» unique ou un groupe limité d'établissements peut être disponible.
Les traitements de chimiothérapie pour les RMS sont toujours administrés par voie intraveineuse. En général, un type spécial de perfusion intraveineuse "permanente" est placée avant le début du traitement. La plupart des patients atteints de RMS reçoivent des traitements de chimiothérapie durant 6 à 12 mois (rarement plus, bien que selon la gravité des effets secondaires, un traitement qui est prévu durer dix mois peut parfois durer 15 mois). La chimiothérapie est généralement administrée en deux à cinq (ou parfois dix) jours par "cycles" tous les trois à quatre semaines. Certains médicaments de chimiothérapie peuvent être donnés toutes les semaines.
Les effets secondaires de la chimiothérapie peuvent être "spécifiques" au médicament (c'est-à-dire, vus avec un ou deux médicaments) ou «globaux» (c'est-à-dire, vus avec de nombreux médicaments). Voici une liste des médicaments les plus communément utilisés pour traiter les RMS aux États-Unis et en Europe:
- Vincristine
- Dactinomycine
- Cyclophosphamide
- Topotécan
- Irinotécan
- Etoposide
- Ifosfamide
- Doxorubicine
- Carboplatine
Les effets indésirables fréquents qui peuvent être vus (à des degrés plus ou moins importants) avec pratiquement tous les médicaments de chimiothérapie qui sont utilisés pour traiter les RMS, comprennent la perte de cheveux, les nausées et vomissements, la perte d'appétit, la fatigue, l'inflammation des muqueuses (mucite) et une diminution du nombre de cellules sanguines. Ces effets secondaires se développent généralement à cause des effets de la chimiothérapie sur les cellules à division rapide. Alors que les cellules tumorales sont habituellement les cellules qui se divisent le plus rapidement dans le corps, d'autres cellules normales, telles que les cellules des cheveux, les cellules de la muqueuse (les cellules qui tapissent la bouche et les intestins) et les cellules sanguines, sont également à division rapide. Heureusement, il y a habituellement une plus grande disponibilité de ces cellules normales que des cellules tumorales de sorte que ces effets secondaires sont généralement temporaires.La diminution du nombre de cellules sanguines est l'effet secondaire qui limite le plus la capacité d'administrer une chimiothérapie en permanence (tel qu'une infection serait traitée) et est l'un des effets secondaires les plus dangereux. Il existe trois grands types de cellules sanguines: globules rouges, globules blancs et plaquettes. En règle générale, environ sept ou huit jours après le début d'un cycle de chimiothérapie, les nombres des cellules sanguines tombent à des niveaux très bas et peuvent rester faibles pendant 5 à 10 jours. Les globules rouges transportent l'oxygène des poumons au reste du corps. On appelle anémie le manque de globules rouges et celle-ci entraîne la fatigue. Les globules blancs sont les cellules qui luttent contre les infections de l'organisme. Lorsque le nombre de globules blancs est faible c'est ce qu'on appelle une leucopénie et cela peut augmenter considérablement le risque de développer une infection grave à partir des «germes» qui sont déjà présents dans / sur son propre corps. Quand les globules blancs les plus important dans le combat des infections sont en nombre faible, cette condition est appelée neutropénie. Les plaquettes sont les cellules qui aident le sang à coaguler. Lorsque le nombre de plaquettes est faible, le risque de saignement, soit spontané, soit à partir d'une coupure, augmente. Lorsque le nombre de globules rouges est faible, une transfusion peut aider à améliorer la fatigue. Lorsque les plaquettes sont basses, une transfusion peut être donnée pour réduire le risque de saignement. La plupart des enfants atteints de RMS, même ceux à risque standard, aux tumeurs du sous-groupe A, qui reçoivent une chimiothérapie à 2 médicaments relativement moins intensive avec la vincristine et la dactinomycine, auront besoin du soutien de transfusions de globules rouges et / ou de plaquettes à un moment donné au cours de leur traitement. Les seules cellules sanguines qui ne peuvent être transfusées sont les globules blancs qui combattent les infections. Toutefois, un médicament (G-CSF, filgrastim, Neupogen ®) est disponible pour aider le nombre de globules blancs à retrouver un niveau sain plus rapidement.
L'inflammation du foie, bien qu'un effet secondaire rare, peut se produire et peut être mortelle, particulièrement chez les enfants très jeunes. Elle nécessite un niveau accru de sensibilisation pour suivre et évaluer sans délai les tests de laboratoire de "la fonction hépatique".
Bien que rare, le développement d'une "hépatopathie" induite par la chimiothérapie est une complication potentiellement mortelle. Cette pathologie est caractérisée par une hyperbilirubinémie, des ascites, des troubles de la coagulation et l'inversion du sens d'écoulement dans la veine porte à l'échographie Doppler. Un âge de moins de trois ans augmente le risque. Des modifications du dosage de la chimiothérapie fondées sur l'âge peuvent réduire le risque d'hépatopathie, en particulier chez les jeunes enfants (réf. 40).
La vincristine est un médicament qui est donné à pratiquement tous les enfants atteints de RMS. Exceptionnellement, il peut provoquer des douleurs dans les mains et les pieds ou dans la mâchoire ou l'abdomen. Il peut aussi produire une faiblesse dans les mains et les pieds (généralement réversible) à cause de lésions au niveau des nerfs (neuropathie périphérique). À l'heure actuelle, il n'existe pas de médicament "de protection" d'efficacité prouvée pour prévenir ces lésions nerveuses mais il existe des preuves que les lésions du système nerveux causées par d'autres médicaments de chimiothérapie (généralement non utilisés pour traiter les RMS), en particulier le cisplatine et le paclitaxel, peuvent être améliorées par l'utilisation de la vitamine E et de la glutamine, respectivement.
La gestion de la neuropathie périphérique associée à la vincristine reste problématique. Bien qu'il n'y ait pas eu d'étude prospective formelle, l'expérience clinique indique que les patients de plus de huit ans tolérent l'utilisation intensive de la vincristine moins bien que les patients plus jeunes. Deux autres médicaments de chimiothérapie couramment utilisés, le cisplatine et le paclitaxel, causent aussi une neuropathie périphérique. Deux études ont montré que l'utilisation concomitante de la glutamine avec le cisplatine (réf. 41) et de la vitamine E avec le paclitaxel (réf. 42), peut réduire l'incidence et la sévérité de la neuropathie périphérique. Bien qu'aucun de ces agents n'ait été formellement évalué chez les enfants atteints de neuropathie périphérique associée à la vincristine, l'expérience clinique anecdotique suggère que les deux sont sans danger, sont bien tolérés et peuvent être utiles dans certains cas.
L'irinotécan est un nouveau médicament qui a été jugé très efficace dans le traitement des RMS pour les patients nouvellement diagnostiqués avec des tumeurs métastatiques et pour les patients atteints de RMS récurrent (c'est-à-dire, victimes d'une rechute après la fin du traitement ou qui n'ont pas connu de rémission après le traitement initial). Exceptionnellement, il est administré pendant dix jours toutes les trois semaines et même s'il provoque seulement occasionnellement des nausées ou des vomissements sévères, une diminutiom du nombre de cellules sanguines ou une perte de cheveux, il peut produire des diarrhées très graves.
L'irinotecan (CPT-11) est un nouveau médicament prometteur avec un taux très élevé d'activité révélé par des études pré-clinique dans un modèle murin de xénogreffe de RMS (réf. 13). Les essais cliniques chez les enfants atteints d'une rechute ont également démontré une très bonne activité (réf. 13, 43). Les études actuelles IRS-V l'utilisent à la fois pour les enfants nouvellement diagnostiqués avec des tumeurs métastatiques (D9802) et pour les enfants atteints de tumeurs récurrentes (ARST0121). Un essai clinique pilote du centre cancéreux américain MSKCC l'utilise également pour les patients atteints de RMS à risque intermédiaire et élevé, à la fois comme un agent cytotoxique "classique" , un agent potentiel de radiosensibilisation et un agent anti-angiogénique potentiel lorsqu'il est administré en "traitement d'entretien." Bien que généralement bien toléré en termes de toxicité plus traditionnelle comme l'alopécie (perte de cheveu), les nausées, les vomissements et la cytopénie, son utilisation est associée à une incidence élevée de diarrhées, potentiellement graves nécessitant une réhydratation intraveineuse. Des directives publiées existent pour la gestion de cette complication (réf. 44).
Le cyclophosphamide (habituellement administré en association avec la vincristine et la dactinomycine ou la vincristine et la doxorubicine) et l'ifosfamide (habituellement administré en association avec l'étoposide) peuvent causer des dommages à la vessie se traduisant par du sang dans l'urine. Les deux médicaments sont administrés avec un médicament de «protection», appelé le "mesna" qui est efficace pour réduire le risque de cet effet secondaire spécifique.
La doxorubicine peut causer des dommages au coeur, en particulier à des doses totales (cumulatives) plus élevées . De plus en plus, pour les RMS et les autres types de cancer, elle est donnée avec un médicament de protection appelé la "dexrazoxane" qui est efficace pour réduire le risque de cette complication potentiellement très grave.
En dépit de son activité anti-tumorale marquée, le développement de lésions cardiaques potentiellement mortelles, même des années après son administration, a été un facteur conduisant à l'élimination de la doxorubicine dans les essais cliniques récents des groupes de coopération pour les patients atteints de RMS. Il a été démontré que l'utilisation de la dexrazoxane permet de réduire de manière significative le risque de dommage cardiaque associé au traitement par la doxorubicine (réf. 45) sans pour autant réduire l'efficacité anti-tumorale de la doxorubicine (réf. 46).
Radiotherapie
Tous les patients atteints de RMS alvéolaires, même ceux dont les tumeurs ont été complètement enlevées avant le début de la chimiothérapie, et presque tous les patients du groupe II (maladie résiduelle microscopique) et du Groupe III (maladie résiduelle visible à l'oeil nu) atteints de RMS embryonnaire, doivent être traités par radiothérapie pour maximiser leur chance de guérir. Les filles atteintes de RMS embryonnaire de l'appareil génital (vagin, vulve, col de l'utérus et utérus), pour qui un traitement chirurgical initial traditionnel est de règle, peuvent souvent être gérées par une série de biopsies, qui commence après environ 12 semaines de traitement par chimiothérapie, sans radiothérapie. La radiothérapie est généralement administrée après 4 à 5 cycles de chimiothérapie (c'est-à-dire après environ 12 semaines) bien que dans certains cas (généralement limités aux enfants atteints d'un RMS paraméningé qui a érodé la base du crâne pour atteindre l'intérieur du crâne) la radiothérapie peut être administrée en même temps que (ou aussitôt que possible après) la chimiothérapie.
Selon l'emplacement, la taille et le groupe de la tumeur, entre 20 et 28 traitements de radiothérapie sont administrés. Idéalement, le traitement doit être envisagé en se fondant sur l'imagerie tridimensionnelle de la tumeur effectuée avant la biopsie et la chimiothérapie. La compétence du radiothérapeute pour le succès du traitement d'un RMS ne peut être surestimée. Parce que ce sont des tumeurs rares et parce que la plupart des enfants atteints d'un RMS sont traités suivant des protocoles qui précisent les modalités de leur thérapie, le radiothérapeute doit non seulement être en mesure d'interpréter correctement résultats d’imagerie pour concevoir une "zone de traitement" approprié qui englobe l'ensemble de la tumeur d'origine plus une marge "de tissu sein" qui l'entoure, mais doit le faire au moment précisé par le protocole et en étant conscient de la « tolérance des tissus normaux » des structures environnantes normales et les risques de complications à long terme de l'irradiation de tissus en plein développement chez un jeune enfant.
Quelques-uns des groupes européens de coopération qui traitent les enfants atteints de RMS ont essayé de réduire ou d'éliminer l'utilisation de la radiothérapie chez les très jeunes enfants ou chez les enfants dont les tumeurs ont disparu complètement après une période de chimiothérapie ou qui ont été visuellement totalement réséquées avant le début de la chimiothérapie. Malheureusement, bien que certains enfants puissent être guéris de cette façon, le risque de rechute est bien plus élevé et il n'est pas clair si les chances de guérison sont par la suite aussi bonnes. Par conséquent, à l'exception des filles atteintes de RMS embryonnaire au niveau des organes génitaux, le rayonnement est recommandé pour tous les patients du groupe III, pour tous les patients du groupe II et pour tous les patients du groupe I atteints de RMS alvéolaire. Le rôle de la radiothérapie au niveau des emplacements de maladie métastatique chez les enfants au stade 4 (ou groupe IV) est moins clair bien que les enfants présentant des métastases pulmonaires qui ont disparues après une chimiothérapie peuvent voir une amélioration de leur pronostic suite à une radiothérapie à faible dose (habituellement huit cures) visant la totalité du poumon (WLI).
Aucune différence n'a été observée dans l'étude IRS-IV entre l'utilisation de la radiothérapie hyperfractionnée et la radiothérapie conventionnelle fractionnée (XRT) (réf. 36). Si la plupart des patients atteints de tumeurs du groupe III parviennent à un contrôle local avec une XRT à dose complète, l'atteinte des ganglions lymphatiques au moment du diagnostic est corrélée à un risque deux fois plus élevé d'échec du traitement local (réf. 47). La même observation a été faite pour les patients atteints de tumeurs du groupe II, où le plus haut risque de récidive locale a été observé chez les patients atteints de maladie résiduelle microscopique et de l'envahissement des ganglions régionaux (Groupe IIC), réf. 48. Tous les patients atteints de RMS alvéolaire, même ceux qui ont bénéficié d’une résection complète de leur tumeur, devraient recevoir une irradiation locale (réf. 49). Les chercheurs européens ont essayé d'éviter ou de limiter l'utilisation de l'irradiation locale chez les patients du groupe II (réf. 50) et groupe III (réf. 51). Les taux de récidive locale plus importants ont été observés avec cette approche. La connaissance du radiothérapeute des directives de traitement pour les enfants atteints de RMS est très importante (réf. 52). L'utilisation de l'imagerie en 3 dimensions et de la radiothérapie conformationnelle ou à modulation d'intensité (des nouvelles techniques prometteuses pour une radiothérapie très ciblées) ont produit des taux supérieurs de contrôle local, en particulier pour les patients aux tumeurs localisées «à risque élevé», telles les larges tumeurs paraméningées avec extension intracrânienne (réf. 53-54). Seules parmi les patients aux tumeurs du groupe II et III, les filles atteintes de tumeurs embryonnaires de l'appareil génital non réséquées n'ont pas besoin de radiothérapie pour le contrôle local. La gestion optimale de ces patients est généralement constituée d'une chirurgie initiale limitée suivie d'une série de biopsies commençant après une période d'environ douze semaines de chimiothérapie et avec une chirurgie ou une radiothérapie définitive après 24 à 30 semaines si il y a persistance tumorale (des rhabdomyoblasts différenciés ne sont généralement pas considérés comme des preuves d'une tumeur active à cet endroit) (réf. 55).
Chirurgie Retardée
Certains enfants atteints de RMS subissent une chirurgie "retardée" après une réduction de la taille de leur tumeur suite à la chimiothérapie. Ce type d'opération permet : sont soit d'éviter la radiothérapie (rare) soit une radiothérapie post-opératoire de plus faible dose (commun) soit de maximiser les chances que la radiothérapie post-opératoire fonctionne de manière efficace (en particulier pour les tumeurs qui étaient très larges au moment du diagnostic). Parfois, un enfant dont la tumeur a été traitée par radiothérapie aura des résultats d'imagerie inquiétants suggérant que la tumeur n'a pas été éradiquée par les rayonnements. Dans ce cas, la chirurgie peut être nécessaire pour éliminer le cancer résiduel qui a survécu à l'irradiation pour tenter de prévenir une récidive locale.
Le rôle de la chirurgie dans la gestion des patients atteints de RMS dépend clairement de l'emplacement de la tumeur. Les résultats sont suggérés meilleurs lorsqu' une chirurgie initiale complète, totale en apparence, ou de réduction de la tumeur est entreprise pour les patients atteints de tumeurs dans des localisations défavorables (réf 56-58). Puisque un essai randomisé de la résection chirurgicale ne sera probablement jamais accompli, il ne sera probablement jamais possible de dire si cette amélioration du résultat est une fonction de la résection chirurgicale en soi, ou si la résécabilité chirurgicale est simplement associée à d'autres facteurs connus pour donner de meilleurs résultats comme la présence d'une tumeur résiduelle visible au moment du réexamen du traitement initial chez les patients qu'on pensait avoir subi une résection initiale "complète", une taille de tumeur plus petite, une tumeur non-invasive, l'absence d'envahissement ganglionnaire, et une meilleure réponse aux chimiothérapies néoadjuvantes. En règle générale, en particulier pour les patients atteints de tumeurs dans des emplacements défavorables, une chirurgie de conservation maximale de la fonction et de l'esthétique est appropriée au moment du diagnostic. Pour les tumeurs qui ne peuvent être réséquée au moment du diagnostic, une chirurgie retardée doit être considérée en particulier si une résection complète ou en apparence totale est considérée probable et peut permettre une réduction significative de la dose de radiothérapie post-opératoire, ou si l'on se préoccupe de la présence de tumeur résiduelle viable après la radiothérapie (réf. 59). Bien que la chirurgie "non-mutilante" a été de principe au cours des deux dernières décennies, en particulier pour les patients souffrant de tumeurs au niveau de la vessie ou de la prostate, un rapport récent met en garde que la conservation de l'organe qui ne serait pas synonyme de fonctionnement normal de l'organe (réf. 60).
Nouveaux Traitements
Le taux de survie suite à une rechute reste mauvais pour la majorité des patients atteints de RMS récurrent. 95% des récidives surviennent dans les trois ans suivant le diagnostic. À l'exception d'un petit groupe de patients à "risque favorable" (environ 20% des patients victimes d'une rechute), dont la survie à 5 ans est proche de 50%, la moitié des patients atteints de RMS récurrent meurent de leur maladie dans l'année suivant leur rechute et 90% des patients meurent dans les cinq ans suivant leur rechute (réf. 61).
De nouvelles thérapies sont absolument nécessaires pour ce groupe de patients.
Une meilleure connaissance des processus critiques à la genèse des rhabdomyosarcomes (réf. 62-64) permet l'apparition de nouvelles pistes de traitements sur des bases biologiques. Des traitements voués à interrompre les interactions critiques entre les récepteurs et ligands des facteurs de croissance, ou de leurs cibles en aval, semblent particulièrement prometteurs. Une voie autocrine de l'IGF-2 joue un rôle dans la croissance des RMS (réf. 65). Une thérapie biologique "intelligente" potentielle est de perturber cette voie. La croissance de xénogreffes de RMS chez une souris "nude" (ayant un système immunitaire inhibé) peut être inhibée par des anticorps monoclonaux dirigés contre le récepteur de l'IGF-1, le récepteur qui lie l'IGF-2 et sert d'intermédiaire à son signal mitogène (réf. 66). Il a été démontré qu'un nouvel anticorps monoclonal qui reconnaît le récepteur humain de IGF-1 empêche la prolifération de la lignée cellulaire d'un RMS stimulée par l'IGF-1 (réf. 67). Des petites molécules inhibitrices de la tyrosine kinase, hautement spécifiques, ciblées à l'encontre de la tyrosine kinase du récepteur de l'IGF-1, ont été synthétisées et il a été démontré qu'elles inhibent la croissance de xénogreffes tumorales, soit seules soit en combinaison avec une chimiothérapie cytotoxique (réf. 68).
Il est connu que les protéines intracellulaires peuvent être transformées et présentées comme des peptides sur la surface de la cellule par le complexe majeur d'histocompatibilité (CMH) de classe I et a suggérée la possibilité que des produits spécifiques au gène mutant de la tumeur peuvent être la cible des lymphocytes T cytotoxiques (réf. 69 à 70). Par exemple, des chercheurs ont montré qu'un peptide dérivé d'une protéine p53 mutante est spécifiquement reconnu par les lymphocytes T cytotoxiques (réf. 71-72). De la même façon, les protéines de fusion spécifiques à la translocation pourraient éventuellement être ciblées par les cellules T cytotoxiques (cellule TC). Plus précisément, la protéine de fusion PAX-FKHR générée par la translocation t (2; 13) (q35; q14) des RMS alvéolaires est une cible potentielle pour des approches thérapeutiques utilisant les lymphocytes T cytotoxiques. Basé sur des études pré-cliniques sur des modèles murins démontrant que des cellules dendritiques (DC) de la moelle osseuse marquées avec des antigènes associés aux tumeurs (TAA) peuvent générer des lymphocytes NK (Natural Killer) ainsi que des lymphocytes T cytotoxiques CD8+ contre les RMS (réf. 73), des études cliniques pilotes utilisant des vaccinations spécifiques de cellules dendritiques marquées avec le peptide de PAX-FKHR sont en cours. Le succès de cette approche dépendra de la capacité des cellules tumorales à présenter un peptide de fusion transformé lié au CMH sur la surface cellulaire. Si cela peut se produire, plusieurs méthodes pourraient alors être considérées pour surmonter les déficits potentiels qui ont permis à la tumeur d'échapper au départ à l'immunité cellulaire (réf. 74-75).
A mesure que des connaissances sont acquises sur la biologie initiale des RMS, des approches thérapeutiques innovantes sont mises au point pour tenter d'exploiter ces "talons d'Achille " des cellules tumorales. En raison de la dépendance des RMS à l'égard de l'IGF-2, de nouveaux médicaments prometteurs ont été développés soit pour bloquer l'interaction du récepteur de l'IGF-1 avec l'IGF-2 soit pour bloquer les effets biologiques en aval qui se produisent après que l'IGF-2 se lie à son récepteur. Ces agents, pas encore cliniquement disponibles, offrent une bonne perspective, en monothérapie ou en combinaison avec la chimiothérapie.
Enfin, en raison de la présence de l'unique gène de «translocation» spécifique aux cellules tumorales des RMS alvéolaires, le potentiel existe d'utiliser des thérapies basées sur le système immunitaire pour reconnaître et détruire les cellules qui contiennent ce gène anormal. Des essais cliniques pilotes sont en cours pour évaluer la capacité de "vacciner" les patients atteints de RMS alvéolaire pour qu'ils développent une immunité contre leurs propres tumeurs. Simultanément, des essais cliniques pilotes sont également en cours pour évaluer la capacité du système immunitaire "génétiquement compatible" d'un frère ou d'une sœur à contrôler la tumeur d'un patient atteint de RMS alvéolaire, suite à une "mini" allogreffe de cellules souches.
Effets Rardifs du Traitement des Rhabdomyosarcomes
Une thérapie qui prend en compte les risques pour les enfants atteints de RMS est conçue de façon à maximiser les chances de guérison, tout en minimisant le développement de complications à court, moyen et long termes. Les effets tardifs liés au traitement peuvent se développer n'importe quand, de plusieurs mois à plusieurs années après la fin du traitement. Les agents individuels de chimiothérapie peuvent avoir des effets toxiques uniques qui peuvent ne se manifester que plusieurs années après la fin du traitement, ou qui peuvent se détériorer petit à petit au fur et à mesure du suivi. Les dommages causés par la radiothérapie, et les complications tardives de la chirurgie, peuvent ne pas apparaître pendant de nombreuses années, en particulier chez les enfants en pleine croissance. Une sélection des complications bien décrites du traitement comprend:
- L'infertilité (associée notamment à l'utilisation d'agents alkylants tels que le cyclophosphamide et l'ifosfamide): le risque de stérilité induite par la chimiothérapie est beaucoup plus grand pour les garçons que pour les filles (réf. 76). Lorsque cela est possible, même pour les garçons sur le point de développement pubertaire, une évaluation doit étudier la possibilité de cryoconserver le sperme (réf. 77). Bien que le sujet d'études approfondies en laboratoire, la cryoconservation du tissu ovarien ou des ovules n'est actuellement pas disponible en routine pour la préservation efficace de la fertilité chez les filles (réf. 78). Heureusement, le risque d'infertilité semble être beaucoup plus faible chez les filles. Pour les filles en cours d'irradiation pelvienne, ou pour les garçons en cours d'irradiation du scrotum, un déplacement chirurgical de la (les) gonade(s) hors du champ de rayonnement peut être utile pour la préservation de la fonction hormonale et / ou de la fertilité.
- Troubles de la vessie: bien qu'une chirurgie conservatrice "non-mutilante" avec une irradiation à dose complète est devenue le traitement de référence pour la préservation de la vessie chez les enfants atteints de RMS au niveau de la vessie ou la prostate, approximativement la moitié des enfants aux vessies «intactes» sont victimes d'un ou plusieurs symptômes de dysfonctionnement de la vessie comprenant l'écoulement goutte à goutte ("dribbling"), l'incontinence et l'énurésie (réf. 60).
- Dommages dus à l'irradiation des structures de la tête et du cou: l'utilisation de la radiothérapie pour traiter des tumeurs provenant des structures de la tête et du cou est souvent inévitable en raison de l'absence de structures environnantes "non-essentielles" qui pourraient être «sacrifiées» si l'exérèse chirurgicale complète était tentée. Des complications bien décrites de la radiothérapie comprennent la formation de cataracte après administration de doses aussi faible que 1000 cGy au niveau du globe oculaire (réf. 79), une croissance faciale asymétrique à la suite de l'arrêt permanent du développement osseux et de la fibrose (cicatrisation) des tissus environnants, des infections chroniques du sinus, un échec de croissance en raison des dommages à l'hypophyse (réf. 80) et des anomalies dentaires complexes et multiples (réf. 81). On ignore si, les techniques d'irradiation plus récentes plus précisément ciblées comme la radiothérapie à intensité modulée (RTIM), permettront de réduire le risque de complications tardives de l'irradiation des structures de la tête et du cou (réf. 53).
- Cancer secondaire: la complication tardive la plus dévastatrice du traitement pour tout type de cancer, et pas seulement pour les RMS, est le développement d'une deuxième forme de cancer. L'utilisation de la chimiothérapie et de la radiothérapie peuvent entraîner le développement de cancers secondaires. Les cancers secondaires associés à la chimiothérapie sont le plus souvent les leucémies (typiquement la leucémie myéloïde aiguë) et peuvent être associés à l'utilisation d'agents alkylants (le cyclophosphamide et l'ifosfamide) et des inhibiteurs de la topoisomérase II (étoposide et doxorubicine). Le risque de leucémie secondaire est, heureusement, assez faible (généralement entre 1 et 2%). La radiothérapie est également associée au développement de cancers secondaires, le plus souvent d'autres sarcomes (soit des os ou des tissus mous). Aux doses de radiothérapie qui sont actuellement utilisées pour traiter les enfants atteints de RMS, le risque de sarcome secondaire est d'environ 5%, mesuré 20 ans après le traitement initial (réf. 82). Contrairement aux cas des leucémies secondaires, qui se développent généralement dans les quatre années suivant le traitement, la plupart des cas de sarcomes secondaires ne se développent qu'après plus de 5 ans suivant la fin du traitement (réf. 82). La contribution des "facteurs de risque génétiques" sous-jacents au développement de cancers induits par le traitement est activement recherchée.
Vingt-deux tumeurs malignes secondaires se sont développées pour les 1770 patients inscrits aux études IRS-I et IRS-II, dont 11 sarcomes des os liés à la radiothérapie et cinq cas de leucémie aiguë non-lymphoblastique à une médiane de sept ans après le traitement (réf. 83). Trois des patients touchés ont eu une neurofibromatose, et les familles de sept autres des patients atteints avaient des antécédents compatibles avec le syndrôme de Li-Fraumeni, ce qui suggère que la prédisposition génétique joue un rôle important dans le développement d'une tumeur maligne secondaire après le traitement d'un RMS. Les premiers résultats de l'étude IRS-III décrivent la survenue précoce de cinq cas de leucémie myéloïde aiguë chez les enfants, ainsi que d'un cas d'ostéosarcome et un cas de syndrome myélodysplasique (réf. 84). Un rapport préliminaire des tumeurs malignes secondaires dans l'étude IRS-IV trouvent 14 cas chez 13 patients à une durée médiane de 3,2 ans après le diagnostic (réf. 85). Une mise à jour plus récente de l'expérience de l'IRS a identifié 67 tumeurs malignes secondaires et 2 tumeurs malignes tertiaire pour les 4367 patients inscrits aux études de l'IRS entre 1972 et 1997 (réf. 86). Seuls sept avaient un syndrome de prédisposition génétique reconnu. L'incidence cumulative de tumeurs malignes secondaires est estimée à 3,5% mesurée 20 ans après le traitement. Les premières inquiétudes concernant un risque accru de leucémie myéloïde aiguë ou de syndrome myélodysplasique chez les patients recevant l'étoposide ne semble pas s'être concrète, cependant, il est prudent de surveiller prospectivement la contribution des antécédents familiaux au risque de développement de tumeurs malignes secondaires liées au traitement (réf . 87).
-
Environ 2/3 de ces enfants ont moins de 10 ans au moment du diagnostic. Le RMS est plus courant chez les enfants de 1 à 4 ans et peu commun chez les enfants de moins d'un an.
-
L'IRM de l'orbite révèle une masse des tissus mous se développant au niveau de l'angle supéro-médial de l'orbite gauche déplaçant le globe oculaire vers l'extérieur et latéralement.
-
L'IRM des sinus montre une masse du tissu mou, large, envahissante, centrée dans la la cavité naso-sinusienne, envahissant les deux orbites et s'étendant intracrâniennement à la base du crâne.
-
L'IRM de la prostate montre une masse de tissu mou, large, sur le côté droit du pelvis, comprimant la paroi postérieure de la vessie et la paroi antérieure du rectum.
-
Les cellules des RMS embryonnaires sont typiquement moins denses et plus fusiformes. L'appartenance à la lignée musculaire squelettique peut ne pas être évidente lors de l'examen de routine sous le microscope. Une confirmation par immunomarquage avec des anticorps ciblant la desmine, la vimentine, l’actine et la myogénine soutient le diagnostic d'un RMS.
-
Les cellules des RMS alvéolaires sont typiquement plus petites et plus rondes et de plus forte densité cellulaire. Du point de vue architectural, elles peuvent sembler s'aligner le long de pseudo-espaces qui rappellent les petits sacs d'air dans les poumons (alvéoles).
-
Figure 8: Sur-expression du facteur de croissance analogue à l'insuline de type 2 (IGF-2) par perte d'hétérozygotie en 11p15.
-
Figure 9: la translocation réciproque entre PAX et FKHR crée un 'oncogène' hybride.









{kind=link}
{kind=link}
{kind=link}
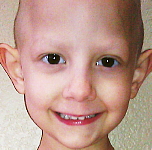{kind=link}
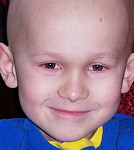{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}