


Rabdomiosarcoma
¿Qué Es el Rabdomiosarcoma?
Existen dos tipos de células musculares en el organismo: células musculares lisas y células musculares estriadas. Las células musculares lisas controlan la actividad involuntaria; las musculares estriadas el movimiento voluntario. El rabdomiosarcoma (RMS) es un tumor maligno ("cáncer") que se origina a partir de células musculares esqueléticas normales. No se sabe con seguridad por qué una célula muscular estriada experimenta transformación neoplásica. Dado que las células musculares esqueléticas están presentes en la práctica totalidad del organismo, el RMS puede desarrollarse casi en cualquier localización.
La primera descripción del rabdomiosarcoma fue realizada por Weber en 1854. Sin embargo, su publicación "definitiva" es atribuida a Stout en 1946, 92 años después.
- Weber, CO. Anatomische Untersuchung Einer Hypertrophieschen Zunge nebst Bemekugen uber die Nubildung querquestreifter Muskelfsern, Virchow Arch. Pathol Anat. 7:115, 1854.
- Stout AP: Rhabdomyosarcoma of the skeletal muscles, Ann Surg 1946; 123: 447-472.

El RMS es una neoplasia muy infrecuente. Se diagnostican sólo unos 350 casos al año en niños de menos de 21 años en los Estados Unidos. Cada año, cuatro niños de entre un millón de niños sanos de edad inferior a 15 años desarrollará un rabdomiosarcoma. Es ligeramente más frecuente en niños que en niñas y la incidencia máxima tiene lugar en niños pequeños (por debajo de 5 años).
El rabdomiosarcoma es muy poco frecuente en adultos. Se han publicado cinco "grandes" series que reúnen un total aproximado de 400 casos de RMS en adultos (incluyendo algunos casos en "niños") valorados en centros oncológicos de referencia en Estados Unidos y Europa a lo largo de los últimos 20-30 años.1-5 Aunque la variante histológica denominada "pleomórfica" es más frecuente en la población adulta (y raras veces es vista en niños), los principios de tratamiento para el manejo de adultos con RMS son similares a los aplicados en niños, y el pronóstico no es intrínsecamente peor para los adultos tratados con terapia multimodal moderna.
Casos en Adultos
 Los principios de tratamiento para el manejo de adultos con RMS son similares que los utilizados en niños. Las cinco series mencionadas más arriba son las siguientes:
Los principios de tratamiento para el manejo de adultos con RMS son similares que los utilizados en niños. Las cinco series mencionadas más arriba son las siguientes:
- Instituto Nazionale Tumori, Milan, Italy, 190 pacientes de 18 o más años de edad durante un periodo de 25 años.1
- 2. Memorial Sloan-Kettering Cancer Center, New York City, NY, 84 pacientes de 16 o más años de edad durante un period de 17 años.2
- 3. M.D. Anderson Cancer Center, Houston, TX, 82 pacientes de 17 o más años de edad durante un periodo de 28 años.3
- 4. Dana-Farber Cancer Institute, Boston, MA, 39 pacientes de 16 o más años de edad durante un periodo de 23 años.4
- 5. Armed Forces Institute of Pathology (Washington, D.C., 38 pacientes de 21 o más años de edad durante un periodo de 30 años, todos ellos con la variante pleomórfica del RMS.5
Los trabajos destacan varios puntos clave en relación al RMS en adultos:
- Muestran una respuesta intrínseca a la quimioterapia tan buena como la del RMS en niños, con tasas de respuesta de hasta el 85%.
- Las variantes histológicas "desfavorables", que incluyen los tipos alveolar y pleomórfico, son más frecuentes que la variante histológica embrionaria (típica de los niños).
- La proporción de pacientes con tumores clasificados en los grupos I, II, II o IV son comparables a los observados en las series pediátricas.
- Con el tratamiento adecuado, incluso considerando las diferencias de proporción de pacientes con variantes histológicas "desfavorables", se obtienen tasas de supervivencia similares a las de las series de pacientes en edad pediátrica.
Aunque estos tumores pueden surgir casi en cualquier órgano, las localizaciones de origen y desarrollo más frecuentes son las estructuras de cabeza y cuello (casi un 40% del total de casos), el tracto genitourinario masculino o femenino (alrededor del 25% de la totalidad) y las extremidades (que suponen aproximadamente un 20% de los casos).
Location |
% |
|---|---|
Parameningeal |
16 |
Orbit |
10 |
Head/Neck |
10 |
GU (all) |
23 |
Extremity |
19 |
Other |
22 |
Aproximadamente el 40% de los nuevos casos de RMS diagnosticados se originan en las estructuras de cabeza y cuello, incluyendo el espacio parameníngeo (16% de la totalidad, y casi la mitad del total de casos originados en cabeza y cuello), la órbita y el párpado (10% del total), y otros lugares extraorbitarios y fuera del espacio parameníngeo (10% de los casos). Alrededor del 25% se originan en estructuras del sistema génito-urinario, incluyendo la región paratesticular, el tracto génito-urinario femenino (vulva, vagina, cérvix, útero), la vejiga urinaria y la próstata. Un 20% aproximadamente aparece en una extremidad. Los casos restantes ("otros") se originan en diversas localizaciones, incluyendo la pared torácica y el retroperitoneo.
Los tumores originados en la órbita, en localizaciones no parameníngeas de cabeza y cuello (por ejemplo, la mejilla o el lóbulo de la oreja) y el tracto genital masculino (espacio paratesticular) o femenino (vagina, vulva, cérvix o útero) son considerados "favorables." Cualquier otro lugar de origen es considerado como "desfavorable."
La mayoría de los niños que desarrollan un RMS no tienen un factor de riesgo evidente para padecer un cáncer. Tras la realización de una minuciosa historia familiar y de una exhaustiva exploración física, aproximadamente uno de cada cinco a uno de cada diez niños tendrá un "factor de riesgo genético" identificable:los más frecuentes de estos "síndromes" genéticos son el síndrome de Li-Fraumeni.6 la neurofibromatosis,7 el síndrome de Beckwith-Wiedemann8 y el síndrome de Costello.9
Aunque la inmensa mayoría de los casos de RMS aparece de forma esporádica, se cree que entre un 10 y un 33% de los niños que desarrollan un RMS tienen un factor de riesgo genético subyacente.10 El desarrollo del RMS se ha asociado con cierto número de raros síndromes de "cáncer familiar" como el syndrome de Li-Fraumeni (SLF), que incluye la tendencia a la agrupación familiar de casos de RSM y otros tumores de partes blandas de niños, con aparición en los familiares adultos de carcinoma córtico-suprarrenal y carcinoma de mama de desarrollo en edades tempranas. El SLF ha sido asociado a mutaciones en la línea germinal del gen supresor tumoral p53.11 En un estudio de 33 casos esporádicos de RMS se encontró que 3 de 13 niños menores de 3 años de edad al diagnóstico (frente a ninguno de los 20 niños restantes, todos ellos mayores de 3 años), tenían mutaciones del gen de p53 en la línea germinal, mientras que ninguno de los 20 niños restantes (todos ellos mayores de 3 años) la presentaban.12 El rabdomiosarcoma se ha observado también asociado al síndrome de Beckwith-Wiedemann, un síndrome de macrosomía (sobrecrecimiento) fetal asociado a anomalías en 11p15, lugar de localización del gen que codifica el factor de crecimiento asociado a insulina tipo II (IGFII).
Los estudios de niños con síndrome de Costello, un desorden autosómico dominante caracterizado por retardo del crecimiento post-natal, rasgos faciales toscos típicos, piel laxa y retraso del desarrollo, han demostrado un riesgo incrementado de desarrollo de tumores sólidos, más frecuentemente rabdomiosarcomas. Se han descrito 10 casos de RMS en el grupo de unos 100 casos conocidos de niños con síndrome de Costello.
Sintomatología del Rabdomiosarcoma
Los síntomas asociados al RSM pueden variar ampliamente dependiendo del lugar en que éste se desarrolle. Los niños con RMS orbitario (alrededor del 10% del total de casos de RMS) pueden presentar un ojo protruyente o hinchado (proptosis). Aunque en ocasiones este cuadro puede ser confundido con una infección del seno orbitario (sinusitis), los niños con tumores en esta localización carecen de los síntomas adicionales que habitualmente acompañan a esta infección (dolor, fiebre, coloración purpúrica del ojo).
Caso 1

Niño de 7 años que se presenta con hinchazón y dolor del ojo izquierdo de una semana de evolución, sin fiebre ni rinorrea purulenta. Se le administraron antibióticos intravenosos a fin de tratar una presunta celulitis periorbitaria. Se realizó examen mediante RMI, que demostró una masa de unos 4 cm en partes blandas de (ver más abajo) originada en la región superomedial de la órbita izquierda que desplazaba el globo ocular en sentido anterior y lateral. Se obtuvo una biopsia de la masa mediante una pequeña incision medial, confirmándose el diagnóstico de rabdomiosarcoma embrionario. No se detectaron metástasis en el TAC de tórax ni en el escáner óseo o en la biopsia de médula ósea. El paciente se encontraba pues en un estadio I, grupo II y fue tratado con éxito mediante quimioterapia VA y radioterapia local con 45Gy.
Los niños con tumores originados en alguna de las localizaciones parameníngeas (que incluyen fundamentalmente los senos paranasales, el oído medio y la parte posterior de la garganta) pueden padecer durante semanas de congestión nasal, en ocasiones con rinorrea; a veces puede incluso ser visible una masa en la amígdala o en la parte posterior de la garganta. A diferencia de las infecciones de senos o de garganta, estos tumores no suelen diseminarse en los ganglios linfáticos cervicales. Si lo hacen, las adenopatías suelen ser indoloras. Si se produce erosión de la base del cráneo, el paciente referirá dolor de cabeza o desarrollará una neuropatía craneal por infiltración o compresión de los nervios craneales afectados.
Caso 2

Niña de 14 años que debutó con una historia de 2 semanas de evolución de proptosis derecha de empeoramiento rápido y "glándulas hinchadas" en el lado derecho del cuello. La RMN mostró una masa multicompartimental de casi 7 cm de diámetro (mostrada más abajo) centrada en la cavidad nasosinusal, que se extendía a la fosa craneal anterior través de la lámina cribiforme. No se observó edema en los lóbulos frontales que sugiriera extensión directa al parénquima cerebral. Se observaron también múltiples ganglios linfáticos aumentados de tamaño en el lado derecho del espacio retrofaríngeo y en la cadena ganglionar látero-cervical derecha. En el examen físico destacaba una llamativa proptosis derecha y oftalmoplejia, con visión conservada. De la narina derecha emergía una masa visible acompañada de linfadenopatías cervicales de consistencia pétrea. La punción aspiración con aguja fina (PAAF) de los ganglios linfáticos cervicales mostró un tumor de células pequeñas redondas y azules sugestivo de RMS. La biopsia de la masa situada en la cavidad nasal demostró el patrón "alveolar" característico del RMS del mismo nombre. Las tinciones inmunohistoquímicas mostraron positividad intensa para desmina, vimentina y miogenina. La técnica de RT-PCR (reacción inversa de la polimerasa) confirmó la existencia de traslocación t(2;13) PAX3-FKHR. No se identificaron células tumorales en la citología del líquido cefalorraquídeo. No se demostraron metástasis en el TAC de tórax, escaner óseo, PET o en la biopsia de médula ósea. El diagnóstico fue de RMS alveolar primario parameníngeo (de probable origen en seno etmoidal) con extensión intracraneal, estadio 3, grupo III. La RMN y el PET de control de respuesta al tratamiento demostraron, inmediatamente después de completar dos ciclos de quimioterapia, desaparición completa del tumor de los lugares en los que era visible. Sin embargo, y a pesar del suministro de quimioterapia adicional y de radioterapia a dosis completa (50.4 Gy) en la localización de origen del tumor y en los ganglios linfáticos afectos, seis meses después del inicio del tratamiento se produjo una recidiva leptomeníngea en el campo de radiación, que resultó fatal.
Los niños con tumores originados en el tracto genitourinario pueden debutar con una masa escrotal indolora (tumores paratesticulares), una masa protruyente "en racimo de uvas" en la vagina (rabdomiosarcoma botrioide), sangre en la orina (en los tumores de vejiga urinaria) o micción frecuente, en ocasiones con ardor miccional o interrupción del chorro de micción. En ocasiones, los tumores que se originan en la glándula prostática (distintos al cáncer de próstata que aparece en pacientes adultos, mucho más frecuente) pueden alcanzar un gran tamaño antes de ser diagnosticados; estos tumores pueden debutar como masas visible en la pelvis o en el abdomen, en ocasiones acompañadas de micción frecuente e imperiosa, a veces con estreñimiento, náuseas y vómitos por compresión del intestino.
Caso 3

Estudiante universitario de 18 años que desarrolló disfunción eréctil, dolor en flanco derecho y abdominal agudo, micción frecuente con interrupción y disminución del calibre del chorro de la orina. Se administraron antibióticos orales sin obtener mejoría. El TAC demostró una masa pélvica de 10 x 6.5 x 7.3 cm originada en la vecindad de la próstata, sin plano de separación con la pared posterior de la vejiga urinaria y de la pared anterior del recto, que obstruía el uréter derecho causando hidronefrosis y se acompañaba de adenopatías ilíacas externas bilaterales e internas izquierdas. La RMN mostró hallazgos similares (mostrados a continuación). En la biopsia con aguja transrectal realizada se obtuvo material correspondiente a un tumor de células pequeñas redondas y azules, de elevada densidad celular, con intensa positividad para desmina, vimentina, actina y miogenina en el estudio con técnicas inmunohistoquímicas y que además presentaba una traslocación t(2,13) PAX3-FKHR en la evaluación mediante PCR. Para resolver la hidronefrosis derecha se llevó a cabo una nefrostomía provisional con tubo de drenaje. No se detectaron metástasis en la TC de tórax, escáner óseo o en la biopsia de médula ósea. El diagnóstico fue de RMS alveolar, estadio 3, grupo IIIA; se inició el suministro de quimioterapia agresiva, que consiguió una respuesta completa del paciente. La function eréctil retornó a la normalidad. Se administró además quimioterapia adicional y una dosis total (50.4 Gy) de radioterapia pélvica; el tratamiento se complicó con el desarrollo de cistitis hemorrágica y enteritis por radiación. El paciente volvió a la Universidad menos de tres meses después de completar los 8 meses de tratamiento y permanece en remisión completa 18 meses después del diagnóstico.
Los tumores originados en las piernas o en los brazos se encuentran habitualmente entre los tipos más agresivos de RMS. Estos tumores pueden crecer rápidamente en el curso de unas pocas semanas, pasando de tener el tamaño de una picadura de mosquito o de una cuenta al de una pelota de fútbol o de un racimo de uvas. Los tumores son habitualmente de consistencia dura, aunque raras veces son dolorosos salvo que se originen cerca de los nervios o los compriman en su crecimiento. Estos tumores son también los que con más frecuencia se diseminan a los ganglios linfáticos vecinos; no es raro que un niño con un RMS en la mano o en el brazo tenga también "glándulas hinchadas" en la axila, o que otro con un RMS originado en el pie o en la pantorrilla las tenga en la ingle.
Caso 4

Niño de 7 años al que cuando se le bañaba se le encontró un "nudo" firme e indoloro en la pantorrilla izquierda. El examen físico confirmó la presencia de una masa pétrea en la pantorrilla con ganglios linfáticos claramente aumentados de tamaño en las regiones poplítea e inguinal. La RMN mostró una gran masa de partes blandas con áreas de hemorragia (mostrada en la imagen) que se extendía en sentido cefálico a través de la fosa poplítea. El TAC de tórax, abdomen y pelvis demostró la existencia de adenopatías inguinales y pélvicas y de adenopatías paraaórticas "sospechosas"; el PET confirmó que se trataba de ganglios hipermetabólicos concordantes con metástasis. La biopsia incisional de la masa de la pantorrilla y del ganglio linfático inguinal reveló que se trataba de un RMS alveolar "clásico"; la técnica de RT-PCR confirmó en el tejido tumoral la existencia de una traslocación "de consenso" PAX-FKHR. No se encontraron metástasis en los pulmones, en los huesos o en la médula ósea. El diagnóstico fue de RMS alveolar de la extremidad con metástasis ganglionares regionales (poplíteas e inguinales) y a distancia (pélvicas y paraaórticas). En la primera semana de tratamiento quimioterápico la masa tumoral de la pantorrilla se había reducido en más del 50% y el estado hipermetabólico de los ganglios linfáticos se había resuelto. El tratamiento continua aún, basado en un protocolo de institución única MSKCCT para pacientes de alto riesgo.
En ocasiones los niños con RMS pueden tener también como síntoma en el momento del diagnóstico episodios de fiebre no explicada. Puede haber o no disminución asociada del apetito. La fatiga y los pequeños traumatismos o roces frecuentes no son síntomas habituales, salvo que el tumor se haya diseminado a la médula ósea.
Factores Pronósticos
Aunque el RMS es considerado globalmente como una única enfermedad, hay diferencias importantes de comportamiento entre un RMS y otro dependiendo de su lugar de origen, su aspecto histológico al microscopio, su tamaño y su posible diseminación a otras localizaciones, así como de la cantidad de tumor residual que queda tras la cirugía inicial y de la edad del paciente en el momento del diagnóstico. Todos ellos se denominan "factores pronósticos". Estos factores describen la "probabilidad estadística" de curación, pero no pueden nunca determinar con absoluta seguridad si un niño en concreto se curará, independientemente de cuán "favorables" o "desfavorables" sean sus factores pronósticos.
La tabla siguiente resume cómo la combinación de localización, tamaño tumoral, estado de los ganglios regionales, metástasis a distancia, edad al diagnóstico e histología se utilizan para diseñar terapia ajustada al riesgo en los pacientes con RMS. La columna denominada "Riesgo" clasifica a los pacientes dentro de uno de los cuatro grupos de riesgo (Bajo-A, Bajo-B, Intermedio y Alto) que son utilizados para asignar el tratamiento adecuado) por el Quinto Estudio Intercentros del Rabdomiosarcoma (IRS-V). El número específico de protocolo es el incluido entre paréntesis con una letra "D" seguida de un número de cuatro dígitos.
D9602 es el protocolo de "bajo riesgo" que consiste en paroximadamente once meses de tratamiento quimioterápico bien con la variante A (quimioterapia con dos fármacos, vincristina y dactinomicina [VA], con o sin radioterapia) o con la B (quimioterapia con tres fármacos, vincristina, dactinomicina y ciclofosfamida [VAC], con radioterapia adicional para la práctica totalidad de los pacientes); D9803 es el protocolo para los casos de "riesgo intermedio" consistente en el suministro de quimioterapia aplicando de forma aleoatoria el tipo A (14 ciclos de VAC) o tipo B (8 ciclos de VAC alternando con 6 ciclos de vincristina y añadiendo topotecan y ciclofosfamida) y suministrando además radioterapia; D9802 es el protocolo de "alto riesgo" que consiste en una "ventana fase II" desarrollado en el Laboratorio Houghton del Centro de Investigación Infantil del St Jude, consistente en la administración de irinotecan según el esquema "diariamente x 5 x 2,"13 bien como fármaco único bien en combinación con vincristina, seguido de ocho ciclos de VAC, cuatro ciclos de de vincristina e irinotecan para aquellos pacientes que respondan al irinotecan, o doce ciclos de quimioterapia con VAC para los pacientes que no respondan al irinotecan, seguida de radioterapia. Se espera completar los distintos estudios IRS-V a finales del año 2004, iniciando en 2005-2006 los nuevos estudios que los continuarán.
Definición de los Grupos de Riesgo del Rabdomiosarcoma:
Favorable = Órbita/párpado, cabeza y cuello (excluyendo parameníngeos), génito-urinarios (no vejiga ni próstata)
Desfavorable = Bladder, prostate, extremidad, parameninge, otros (tronco, retroperitoneo, etc)
a = Tamaño tumoral <= cinco centímetros de diámetro.
b = Tamaño tumoral > cinco centímetros de diámetro
EMB = Variantes embrionaria, botrioide o fusiforme o ectomesenquimomas con rasgos embrionarios
ALV = Sarcomas alveolares o indiferenciados, o ectomesenquimomas con rasgos alveolares
N0 = Ganglios linfáticos regionales no afectados
N1 = Ganglios linfáticos regionales clínicamente afectados
NX = Estado ganglionar desconocido
| Riesgo | Estadio | Grupo | Localización | Tamaño | Edad | Histología | Metástasis | Ganglios linfáticos |
|---|---|---|---|---|---|---|---|---|
| Bajo A (D9602) | 1 | I | favorable | a o b |
<21 | EMB | M0 | N0 o N1 o NX |
| 1 | II | favorable | a o b | <21 | EMB | M0 | N0 o NX | |
| 1 | III | solo órbita | a o b | <21 | EMB | M0 | N0 o NX | |
| 2 | I | desfavorable | a | <21 | EMB | M0 | N0 o NX | |
| Low B (D9602) | 1 | II | favorable | a o b | <21 | EMB | M0 | N1 |
| 1 | III | solo órbita | a o b | <21 | EMB | M0 | N1 | |
| 1 | III | favorable (excluyendo la órbita) | a o b | <21 | EMB | M0 | N0 o N1 o NX | |
| 2 | II | desfavorable | a | <21 | EMB | M0 | N0 o NX | |
| 3 | I or II | desfavorable | a | <21 | EMB | M0 | N1 | |
| 3 | I or II | desfavorable | a | <21 | EMB | M0 | N0 o N1 o NX | |
| Intermediate (D9803) | 2 | III | desfavorable | a | <21 | EMB | M0 | N0 o NX |
| 3 | III | desfavorable | a | <21 | EMB | M0 | N1 | |
| 3 | III | desfavorable | b | <21 | EMB | M0 | N0 o N1 o NX | |
| 1 or 2 or 3 | I or II or III | favorable o desfavorable | a o b | <21 | ALV | M0 | N0 o N1 o NX | |
| 4 | IV | favorable o desfavorable | a o b | <10 | EMB | M1 | N0 o N1 o NX | |
| High (D9802) | 4 | IV | favorable o desfavorable | a o b | >=10 | EMB | M1 | N0 o N1 o NX |
| 4 | IV | favorable o desfavorable | a o b | <21 | ALV | M1 | N0 o N1 o NX |
Los oncólogos utilizan un conjunto especial de abreviaturas para describir estos factores. Para niños con RMS, hay dos grupos terminológicos utilizados en la descripción de estos factores. Uno se denomina estadio y el otro grupo clínico (o "grupo", para abreviar). El estadio del RMS depende de tres factores:
- La localización de origen del tumor.
- El tamaño del tumor.
- La presencia o no de diseminación regional o a distancia (ver más abajo).
El Grupo en el que se incluye el RMS depende de la cantidad de tumor residual que queda tras la cirugía inicial. Hay cuatro estadios (estadio 1, 2, 3, y 4) y cuatro grupos (grupos I, II, III y IV). Basándose en la combinación de estos factores, a cada paciente con RMS se le asigna un grupo.
Las tablas incluidas a continuación contienen, de forma detallada, el sistema de estadificación TNM modificado según localización y el sistema de clasificación según Grupo Clínico quirúrgico-patológico utilizado para dividir en categorías a los pacientes con RMS. Estos "sistemas abreviados" constituyen uno de los aspectos más complejos en el tratamiento de los niños con RMS. Cualquier tumor originado en una de las localizaciones consideradas favorables es estadio 1 siempre y cuando no muestre diseminación visible a algún otro órgano "distante" del cuerpo (ver más abajo). Cualquier tumor con evidencia visible de extensión a distancia a algún órgano es siempre estadio IV. Los tumores originados en cualquiera de las localizaciones desfavorable serán estadio 2 (en el caso de que sean "pequeños" y no se hayan diseminado a los ganglios linfáticos) o estadio 3 (si son "grandes" o se han extendido a los ganglios). La mayoría de los niños con RMS tienen tumores en estadios 2 o 3. Dado que el sistema de estadificación TNM no requiere confirmación histopatológica de las anomalías detectadas con técnicas de imagen, pueden surgir problemas relativos a la correcta estadificación de los pacientes; así, por ejemplo, un paciente puede ser clasificado como estadio IV basándose en la presencia de un nódulo pulmonar en el TAC interpretado como metástasis que, tras su extirpación quirúrgica y estudio histopatológico posterior, resulta no contener tejido tumoral.
| Estadio | Localización | T Status | Tamaño | Status ganglionar | Metástasis |
|---|---|---|---|---|---|
| 1 | Favorable | T1 o T2 | a o b | N0, N1, o NX | M0 |
| 2 | Desfavorable | T1 o T2 | a | N0 o NX | M0 |
| 3 | Desfavorable | T1 o T2 | a | N1 | M0 |
| 3 | Desfavorable | T1 r T2 | b | N0, N1, o NX | M0 |
| 4 | Favorable or desfavorable | T1 o T2 | a o b | N0 o N1 | M1 |
Cualquier tumor extirpado de forma completa en la intervención quirúrgica inicial se considera grupo I. Un tumor con evidencia de diseminación a distancia en otro u otros órganos es siempre grupo IV. Un tumor aún visible (mediante escaner o examen físico) tras la cirugía inicial se incluye en el grupo III. En el grupo II se incluyen los tumores que macroscópicamente han sido resecados en su totalidad en la primera cirugía pero en los que quedan aún agregados microscópicos de células tumorales residuales no extirpados, con o sin diseminación a los ganglios linfáticos (siempre y cuando éstos también hayan sido resecados). La mitad de los niños con RMS tienen tumores pertenecientes al grupo III.
| Grupo clínico | Definición |
|---|---|
| I | Resección completa, márgenes (-) |
| IIa | Resección completa, márgenes (+) |
| IIb | Resección completa,márgenes (-),ganglios linfáticos extirpados (+) |
| IIc | Resección completa,márgenes (+),ganglios linfáticos extirpados (+) |
| III | Enfermedad residual macroscópica (incluyendo ganglios linfáticos no resecados) |
| IV | Metástasis a distancia |
Patrones de Diseminación
El RMS puede diseminarse de forma local, regional o a distancia.
- "Extensión local" significa que el tumor infiltra o invade los tejidos situados en la inmediata vecindad del lugar en el que se originó.
- "Extensión regional" significa que el tumor ha migrado a los ganglios linfáticos que drenan la zona en la que el tumor se originó. La probabilidad más alta de diseminación a los ganglios linfáticos se da en niños con tumores originados en las extremidades y en niños mayores (de 10 años de edad o más) con tumores paratesticulares.
- "Diseminación a distancia" significa que el tumor ha viajado a través de la corriente sanguínea a otra parte del cuerpo. Los lugares más habituales de diseminación a distancia del RM son los pulmones, los huesos y la médula ósea.
Es muy infrecuente la diseminación del RMS al cerebro o a otros órganos como el hígado o el bazo. La evidencia de diseminación tumoral en órganos situados a distancia del lugar de origen se denomina "metástasis". Sólo uno de cada cinco niños con RMS desarrollará metástasis a distancia.
Para evaluar el tumor primario y buscar signos de diseminación a otras zonas del organismo son necesarias diferentes pruebas. Las primeras serán siempre una historia clínica cuidadosa y una exploración física. Po lo general, la mejor técnica de imagen para evaluar el tumor primario es la RMN. Proporciona una imagen tridimensional y con frecuencia es de ayuda en la planificación de la radioterapia o la cirugía. El TAC de tórax se realiza de manera rutinaria para evaluar la posibilidad de metástasis pulmonares. Dependiendo de la localización del tumor primario, y a fin de buscar una posible diseminación a los ganglios linfáticos, puede también ser necesario realizar TAC abdominopélvico. El escaner óseo es una prueba de medicina nuclear que examina el esqueleto en su totalidad para determinar si existe diseminación del tumor a los huesos. Otra prueba de medicina nuclear que se utiliza cada vez con más frecuencia es la denominada PET scan (Tomografía de emisión positrónica). Esta prueba es en cierto sentido única, pues visualiza la totalidad del organismo, incluyendo hueso y partes blandas, con lo que puede ser utilizada para clarificar un hallazgo ambiguo en el TAC o en la RMN y también como control de respuesta al tratamiento. Dado que el RMS puede diseminarse a la médula ósea, se practicarán aspirados y biopsias de ésta; se introduce una aguja en el hueso de la cadera y se extrae una muestra de médula ósea que será posteriormente analizada. Estas pruebas se llevan a cabo casi siempre al mismo tiempo que se suministra la anestesia para biopsiar el tumor o realizar la inserción de un catéter venoso central (CVC). A los pacientes con tumores originados en localizaciones parameníngeas se les practicará siempre una punción lumbar con objeto de obtener una muestra de líquido cefalorraquídeo (LCR) que será examinado a fin de asegurarse de que los tejidos de revestimiento del cerebro no han sido infiltrados por el RMS.
Alrededor de un 20% de los pacientes diagnosticados de novo tendrán metástasis a distancia en una o más localizaciones. Un número desproporcionado de ellos presentará la variante histológica alveolar del RMS. De un total de 127 pacientes con RMS metastásico tratado según protocolos IRS-IV, el 46% tenía el subtipo histológico alveolar, frente al 22% de los cerca de 900 pacientes con tumores no metastásicos tratados con el mismo protocolo.14 Casi el 60% de ellos tenía metástasis en una única localización, siendo los pulmones (39%) el órgano más frecuente, seguido de la médula ósea (32%), los ganglios linfáticos (30%) y los huesos (27%). Aunque los aspirados y biopsias de la médula ósea son recomendados de rutina como parte de la evaluación para la estadificación de los pacientes con RMS de novo (con diagnóstico de sospecha o confirmado), la afectación aislada de la médula ósea se encontró únicamente en 12 de 900 pacientes sin otras metástasis conocidas; por tanto, el "rendimiento" de los aspirados y biopsias de médula ósea en pacientes con rabdomiosarcoma "localizado" es inferior al 2%.
Tratamiento del Rabdomiosarcoma. Principios Generales
Todos los niños con RMS son tratados con quimioterapia. La mayoría de los niños recibirá además alguna combinación de radioterapia y cirugía, que dependerá del tamaño y la localización del tumor primario y de la proporción del mismo que puede ser extirpada quirúrgicamente.
Cirugía del Rabdomiosarcoma
El diagnóstico de RMS nunca debe realizarse sin obtener previamente una muestra del tumor a fin de ser examinada en el laboratorio. El procedimiento inicial empleado para obtener la muestra se denomina biopsia. La biopsia se considera una intervención "pequeña"; la mayoría de las veces no requiere pernocta en el hospital. Hay diferentes formas de obtener una muestra tumoral o biopsia:
- Biopsia percutánea con aguja: en este procedimiento la aguja se introduce en el tumor a través de la piel, aspirando y extrayendo en el interior de la aguja un pequeño fragmento del mismo. En ocasiones esta técnica es realizada bajo control de imagen con ultrasonidos o TAC, que guían a la persona que realiza la biopsia. El procedimiento no se hace habitualmente con anesthesia, aunque dependiendo de la localización del tumor y de la edad del niño puede ser necesaria la sedación por vía intravenosa. El que esta técnica sea o no más segura que las descritas a continuación depende de la localización del tumor. La biopsia con aguja es capaz de proporcionar una muestra adecuada para realizar un diagnóstico correcto en aproximadamente el 90% de los casos.
- Biopsia incisional abierta: en este procedimiento, que se realiza casi siempre bajo anestesia, se practica una pequeña incisión en la piel a través de la cual se extrae un pequeño fragmento del tumor. Este procedimiento proporciona una muestra adecuada para emitir un diagnóstico correcto en aproximadamente el 100% de los casos.
- Biopsia excisional abierta: en este procedimiento, que se realiza casi siempre bajo anestesia, se practica una incisión cutánea para tratar de extirpar el tumor en su totalidad. Es una operación de mayor envergadura que cualquiera de las dos anteriores. Esta intervención es adecuada en niños cuyos tumores han sido visualizados en su totalidad, siempre y cuando el cirujano crea que la extirpación completa del mismo es posible sin dejar como secuela un déficit funcional (es decir, si un tumor en la pantorrilla puede ser extirpado sin necesidad de realizar una amputación o cualquier otro procedimiento que comprometa la deambulación) o un defecto estético (esto es, si la resección de un tumor en los senos paranasales puede ser efectuada sin producir una gran cicatriz o deformidad facial.
Dado que los estudios de imagen pueden en muchos casos no ser capaces de detectar diseminación tumoral en los ganglios linfáticos regionales, la evaluación quirúrgica de éstos es obligada en dos situaciones concretas: niños con RMS de las extremidades y niños de 10 o más años de edad con tumores paratesticulares. En el primer caso, si se trata de tumores en la extremidad inferior, deberá realizarse muestreo quirúrgico de los ganglios linfáticos localizados detrás de la rodilla o en la ingle; si el tumor se localiza en la extremidad superior, el muestreo se realizará en los ganglios situados detrás del codo o en la axila.15 El papel de la linfoescintigrafía como técnica de identificación del ganglio centinela está siendo aún investigado. El escáner con PET puede ser de ayuda en la identificación de ganglios sospechosos que no se visualizan con claridad mediante técnicas de imagen convencionales como el TAC o la RMN.
En los casos de niños con tumores paratesticulares, y preferiblemente de forma simultánea a la extirpación del tumor primario (que se realiza mediante una incisión en la ingle similar a la practicada en la cirugía de una hernia, resecando y extrayendo del saco escrotal al mismo tiempo el tumor y el testículo contiguo), debe realizarse evaluación quirúrgica de los ganglios linfáticos retroperitoneales ipsilaterales (es decir, de los localizados en el mismo lado que el tumor). Este último procedimiento se está realizando cada vez con más frecuencia mediante laparoscopia, acelerando la recuperación post-operatoria y, en consecuencia, acortando el tiempo necesario para poder iniciar el tratamiento quimioterápico.16
Es importante recordar que la cirugía por sí sola no es nunca curativa en los niños con RMS. También es importante tener presente que el papel de la cirugía depende estrechamente de la localización del tumor. Aunque la extirpación quirúrgica completa de un tumor originado en una extremidad o en la pelvis puede inicialmente mejorar las posibilidades de curación, la resección completa de un tumor en la órbita o en la vagina casi nunca es necesaria (siendo además casi siempre desaconsejable) para conseguir una muy alta tasa de curación. A pesar de que la mayoría de las familias de niños con sospecha de rabdomiosarcoma desean que el tumor les sea extirpado tan pronto como sea posible, la cirugía inicial casi nunca es una emergencia vital; en todos los casos en los que exista un posible RMS es imprescindible realizar un estudio exhaustivo y completo del tumor mediante técnicas de imagen antes de realizar una biopsia. La incorrecta visualización del tumor de forma previa a la toma biópsica puede desembocar en la pérdida irreversible de la posibilidad de planificar adecuadamente un tratamiento radioterápico de importancia crítica.17
Así mismo, es importante asegurarse de que la biopsia será estudiada de forma adecuada por un patólogo experimentado, que la procesará de la manera idónea para asegurarse de que todos los estudios complementarios requeridos sean realizados en el tiempo y la forma requeridos.
Anatomía Patológica del Rabdomiosarcoma

Una vez biopsiado, el tumor es examinado al microscopio en el laboratorio. La característica definitoria del RMS es la evidencia demostrable de su estirpe muscular esquelética, ya sea por su aspecto microscópico o por el patrón de tinción inmunohistoquímica (inmunotinción). Hay dos clases básicas de RMS: el embrionario y el alveolar. Aproximadamente dos tercios de los niños con RMS tienen el tipo embrionario, más frecuente (o sus variantes fusiforme o botrioide). Estos tumores son más comunes en niños pequeños, particularmente en aquéllos que desarrollan tumores en cabeza y cuello (incluyendo la localización parameníngea) y en el sistema genitourinario (incluyendo vejiga y próstata). Las células tumorales tienden a ser más elongadas y el tumor muestra una apariencia menos celular.

Alrededor de un 20-25% de los niños con RMS tienen la variante alveolar, menos frecuente (también denominada variante alveolar sólida). Estos tumores son mucho más frecuentes en adolescentes y asientan de forma habitual en las extremidades. Las células tumorales tienden a ser más pequeñas y redondas, ofreciendo un aspecto densamente celular, y deben su denominación a la similitud que muestran con los pequeños espacios aéreos saculares del pulmón (los alveolos pulmonares). Los tumores alveolares son considerados con frecuencia más "agresivos" o de más "alto riesgo" que las formas embrionarias, especialmente aquéllas que se originan en alguna de las localizaciones consideradas "favorables".
Un 5-10% de los niños con RMS presenta tumores que no pueden ser tipificados o categorizados de manera definitiva y que son considerados bien rabdomiosarcomas "indiferenciados", bien "rabdomiosarcomas, no específicos."
Cuando un tumor ha sido biopsiado y el patólogo (el médico que estudia la neoplasia en el laboratorio) sospecha que se trata de un RMS, éste solicitará habitualmente tests de confirmación denominados "inmunotinciones" (técnicas de inmunohistoquímica). Se trata de reacciones químicas que tiñen diferentes estructuras en las células tumorales. El RMS habitualmente se tiñe (es decir, es "positivo") con cierto número de marcadores entre los que se incluyen la desmina y la miogenina. La demostración de positividad para la miogenina es virtualmente diagnóstica de RMS.

En ocasiones se realiza en las células tumorales del RMS un último test, de nivel más avanzado. Se denomina "test de diagnóstico molecular". Aunque no se sabe demasiado sobre las causas por las que una célula muscular esquelética se transforma en célula cancerosa, hay un amplio conocimiento de los cambios genéticos que tienen lugar en una célula una vez que se convierte en célula tumoral. En la práctica totalidad de los casos de RMS embrionario puede encontrarse en las células tumorales (y sólo en las células tumorales, luego ¡no es una alteración hereditaria!) una anomalía que provoca una "sobredosis" de un gen de gran trascendencia en el crecimiento de las células musculares normales.
Los casos de RMS embrionario muestran de forma característica evidencia de sobreexpresión del gen IGF-II localizado en el brazo corto del cromosoma 11. Se cree que ésta es el resultado de la pérdida del alelo materno y de la duplicación de alelo paterno. Parece ser que la expresión de las dos copias de este gen llevaría a un efecto de "sobredosis" en la que el exceso de IGF-II produciría una señal constante de inducción a la proliferación que permitiría a las células musculares preneoplásicas (o ya con transformación tumoral) crecer de forma incontrolada, sin morir tras exponerse a condiciones ambientales de stress que en otra circunstancia serían letales. Este proceso es conocido como "pérdida de heterocigosidad".

El fenómeno resulta en una "sobredosificación" de un gen promotor del crecimiento denominado factor de crecimiento análogo a la insulina tipo II (IGF-II), que se localiza en el cromosoma 11. Normalmente solo una copia de este gen (habitualmente la del gen heredado del padre) es "activo", mientras que el otro permanece "silente" (se cree que una modificación química de la estructura del DNA próxima al gen, denominada "metilación", es la responsable de activar un gen y de desactivar otro gen supresor del crecimiento [H19] situado en su proximidad). En la mayoría de los RMS embrionarios, cualquiera de los dos genes está activado o bien se pierde la copia del gen materno y el paterno se duplica, siendo mabas copias "activas". Se cree que ello lleva a una señal de estímulo a la proliferación constante, que induce a la célula a continuar creciendo y que evita su muerte en respuesta al stress ambiental al que en condiciones normales ha de enfrentarse.
Muchos de los casos de cáncer en niños están asociados a translocaciones específicas en las que parte de un gen normal y un fragmento de otro gen también normal se separan e intercambian. En aproximadamente el 90% de los casos de RMS alveolar, parte de uno de los genes PAX (con mayor frecuencia el gen PAX3 localizado en el cromosoma 2 y menos frecuentemente el gen PAX 7 situado en el cromosoma 1) se fusiona con una porción del gen FKHR (localizado en el cromosoma 13) creando un nuevo gen "híbrido" (PAX-FKHR) que activa genes de estimulación del crecimiento que en otras circunstancias permanecerían "silentes" e inactiva genes de inhibición del crecimiento habitualmente activos. Dado que este gen híbrido anormal se detecta únicamente en casos de RMS alveolar, su identificación es de gran utilidad en el diagnóstico, pudiendo ser además utilizado en el futuro como diana potencial en inmunoterapia. Esta anomalía cromosómica puede ser detectada mediante alguna de las distintas técnicas especializadas que estudian el contenido cromosómico de las células tumorales.
Cerca del 90% de casos de RMS alveolar tendrá una traslocación característica que involucra uno de los genes PAX (más comúnmente el gen PAX 3, localizado en el cromosoma 2, con menos frecuencia el gen PAX 7, situado en el cromosoma 1) y el gen "forkhead" (FKHR) localizado en el cromosoma 13.
La traslocación es un evento bastante común en los tumores pediátricos en la que parte de un gen normal se fragmenta y separa de su localización habitual uniéndose a parte de otro gen normal. De manera específica, mediante la fusión de PB y HD (dominio de unión) de las regiones de unión del DNA del gen PAX 3 con el "dominio de activación de la transcripción" (TAD) del gen FKHR, se crea un nuevo gen "híbrido" que parece desempeñar un papel crítico en el proceso mediante el que, a través de dos vías, las células del RMS se vuelven cancerígenas. En primer lugar, "inactiva" otros genes que son normalmete activos y que funcionan como "frenos" del crecimiento celular; en segundo lugar, "activa" otros genes que son normalmente incativos y que actúan como estimuladores del crecimiento, supervivencia y diseminación celular. Esta anomalía no se observa en el tipo embrionario de RMS, de modo que si existe alguna duda sobre el tipo de RMS que presenta un paciente por su aspecto histológico, la detección de la traslocación demuestra de forma concluyente que se trata de un RMS alveolar. Esta alteración se estudia habitualmente mediante una técnica denominada RT-PCR (transcriptasa inversa de la reacción en cadena de la polimerasa). No obstante, esta prueba puede estar disponible únicamente en laboratorios especializados de referencia en grandes hospitales pediátricos o centros oncológicos.
Los dos principales subtipos histológicos de RMS, ésto es el embrionario y el alveolar, parecen tener alteraciones genéticas características, diferentes entre sí, que presumiblemente jueguen un papel en la patogénesis de estos tumores. El RMS alveolar ha demostrado poseer una traslocación característica entre el brazo largo del cromosoma 2 y el brazo largo del cromosoma 13, que de forma abreviada se refiere como t(2;13)(q35;q14).18-19 Esta traslocación ha sido clonada molecularmente, demostrando que involucra la zona de yuxtaposición del gen PAX3 (o bien, más raramente, el gen PAX7 localizado en 1p36), considerado un regulador de la transcripción durante el desarrollo muscular precoz, y el gen FKHR, también conocido como FOXO1a, un miembro de la familia de factores de transcripción.20-21 Presumiblemente, el resultado de la fusión de esos factores de transcripción es la activación anormal de la transcripción de uno o varios genes que contribuyen a la transformación fenotípica. Aunque la consecuencia precisa de esta traslocación, específica de este tumor, ha de ser aún dilucidada, utilizando técnicas de análisis de microarrays de cDNA es ha demostrado que la fusión PAX-FKHR expresada en fibroblastos activa específicamente un conjunto de factores miogénicos.22 Más aún, se ha observado que PAX-3-FKHR incrementa la expresión de c-MET, un receptor tirosín-kinasa implicado en la transformación tumoral.23 La técnica de PCR (reacción en cadena de la polimerasa), basada en el estudio de los genes, tenderá a utilizarse de forma progresivamente más amplia en un futuro próximo para la confirmación fidedigna del diagnóstico del RMS. Recientemente se ha identificado, en aproximadamente el 20% de los casos de RMS alveolar, una nueva amplificación en 13q31; este hecho sugiere que uno o más genes de este locus contribuirían a la patogenia de esta neoplasia.24
Se sabe que el otro subtipo histológico principal, el RMS embrionario, muestra pérdida de heterozigosidad (LOH) en el locus 11p15.25-26 Más aún, se ha demostrado que esta LOH conlleva pérdida de material genético materno y duplicación del paterno en el locus citado.27 Esta región es de especial interés dado que aquí se sitúa el gen IGF-II, que codifica un factor de crecimiento del que se cree que juega un papel en la patogenia del RMS (ver discusión posterior). IGF –II sufre un fenómeno de "imprinting" por el cual sólo el alelo paterno es transcripcionalmente activo.28-29 Es por consiguiente concebible que en este tumor el fenómeno de LOH en asociación a la disomía paterna conduzca a la sobreexpresión de IGF-II. No obstante, es también posible que LOH en 11p15 refleje quizá la pérdida de actividad de un supresor tumoral aún no identificado, o que tanto la activación de IGF-II como la pérdida de actividad supresora tumoral sean ambas resultado de la LOH en 11p15 en los casos de RMS embrionario.30
Varios investigadores han publicado recientemente los hallazgos obtenidos mediante el análisis de estudios de hibridación genómica comparada (CGH) entre las células del RMS y líneas celulares. Entre estos hallazgos hay tres muy destacados. Primero, las regiones con amplificación genética se han detectado en las variantes alveolar y embrionaria anaplásica de RMS, sugiriendo que estos subtipos comparten similares eventos genéticos.31 En segundo lugar, varios estudios han evidenciado la existencia de amplificación significativa del locus 15q25-26, lugar donde se codifica el receptor de IGF-I24,31 confirmándose además mediante amplificación específica de IGF-I R mediante PCR y FISH.31 Esto es de especial relevancia, dado que las señales mediadas por IGF están implicadas en la patogenia del RMS. Por último, dos estudios han demostrado pérdida de 9q22 en aproximadamente el 33% de los tumores. Esta región corresponde al locus PTH, un gen supresor tumoral implicado en el desarrollo del RMS en un modelo animal (ratones) del síndrome de Gorlin.31-32
Una vez que se han completado todos los estudios de imagen y que se ha realizado la toma de biopsia confirmándose el diagnóstico de RMS, es ya posible clasificar a los pacientes con RMS en uno de los cuatro "grupos de riesgo" basados en la combinación del estadio (localización, tamaño, afectación ganglionar), grupo (magnitud del tumor residual tras la cirugía), edad al diagnóstico, subtipo histológico (embrionario frente a alveolar) y la presencia o ausencia de metástasis a distancia. Estos grupos de riesgo proporcionan importante información sobre la probabilidad de curación en cada caso con el uso de tratamientos de mayor o menor intensidad:
- Riesgo estandar, subgrupo A: Estos pacientes, relativamente escasos en número, tienen una supervivencia mayor del 85% con protocolos de quimioterapia no excesivamente intensos, de dos fármacos, con o sin radioterapia. Este grupo está constituido fundamentalmente por pacientes con tumores orbitarios (siempre y cuando no hayan metastatizado), pacientes con tumores de localización "favorable" (estadio 1) que hayan sido extirpados por completo durante la cirugía (grupo I), o resecados macroscópicamente en su totalidad dejando únicamente tumor microscópico residual (grupo II), así como pacientes con tumores de localización desfavorable (estadio 2) extirpados en su totalidad (grupo I).
- Riesgo estandar, subgrupo B: Estos pacientes, ligeramente más numerosos, tienen una supervivencia superior al 80% pero requieren una quimioterapia más intensiva, con el uso de 3 fármacos, complementada además con radioterapia (con una excepción importante, ver más abajo). Este grupo está compuesto por: 1) todos aquéllos pacientes con tumores no metastásicos de localización favorable no orbitaria (estadio 1) que son aún visibles (grupo III) tras la cirugía inicial; 2) pacientes con tumores pequeños no metastásicos en localizaciones desfavorables, sin diseminación a los ganglios linfáticos regionales (estadio 2) que han sido macroscópicamente resecados por completo (grupo II) y 3) pacientes con tumores en localizaciones desfavorables que son de gran tamaño o afectan a los ganglios linfáticos regionales (estadio 3) pero que han sido resecados bien por completo bien macroscópicamente (grupos I y II). Aproximadamente un 15-10% de los RMS diagnosticados de novo serán considerados de "riesgo estándar". Los pacientes con RMS alveolar nunca son considerados de bajo riesgo.
- Riesgo intermedio: Estos pacientes constituyen la mayoría de los RMS diagnosticados de novo e incluyen aquéllos con tumores de localización desfavorable (estadios 2 y 3) que no han sido resecados por completo (grupo III), pacientes de edad inferior a 10 años con RMS embrionario diseminado a otras partes del organismo (estadio IV, grupo IV) y todos los pacientes con RMS alveolar no metastásico. Aunque es un grupo heterogéneo de pacientes, la probabilidad de curación con 3 o más fármacos quimioterápicos y radioterapia es habitualmente superior al 50%, llegando en determinados subgrupos incluso a porcentajes tan altos como el 70%.
- Alto riesgo: Estos pacientes suponen aproximadamente un 10-15% de los pacientes con RMS diagnosticado de novo. La probabilidad de curación de estos niños es en general bastante pobre, habitualmente de entre el 20% y el 35%, incluso tras quimioterapia muy agresiva, radiación y cirugía. Este grupo incluye todos los pacientes con RMS alveolar, pacientes de 10 o más años de edad con RMS embrionario metastásico y probablemente dos grupos más habitualmente considerados de riesgo intermedio: niño de menos de 1 año de edad con RMS embrionario metastásico, para los que la tasa de supervivencia a los 5 años es inferior al 20%33 y niños con tumores en las extremidades que tienen diseminación ganglionar, casi todos los cuales presentan la variante alveolar del RMS, para los que la supervivencia a los 5 años es del 30% aproximadamente.15
Tratamiento del Rabdomiosarcoma
El tratamiento de los pacientes con RMS es multidisciplinar y comienza incluso antes del inicio real del mismo con la disponibilidad y participación de radiólogos expertos capaces de interpretar con rigor y exactitud los resultados de las pruebas de imagen, patólogos bien entrenados y familiarizados con el muestreo y la evaluación de los tumores pediátricos de "células pequeñas redondas y azules" y cirujanos hábiles que comprendan el papel de la cirugía inicial en el manejo global de los pacientes con RMS. Incluye también la participación de oncólogos radioterapeutas y oncólogos pediátricos familiarizados con las guías de tratamiento nacionales o institucionales, también conocidas como "protocolos", para tratar esta rara forma de cáncer. De modo óptimo el tratamiento se llevará a cabo en centros donde de forma periódica se lleven a cabo reuniones de especialistas en todas estas materias (denominadas "Comités de Tumores"), de manera que todos los profesionales de la salud implicados en el cuidado del niño tengan acceso a las pruebas de imagen, resultados de las biopsias y evaluaciones durante el tratamiento necesarias para proporcionar una atención óptima.
Dada la corta edad de estos pacientes el equipo de tratamiento debe de incluir también anestesistas para sedar a los niños durante la realización del escaner u otros procedimientos (incluyendo en ocasiones las 5-6 semanas completas que dura el tratamiento con radioterapia), y personal de enfermería familiarizado con las necesidades médicas y complicaciones de los niños con cáncer. Por último, se ha de incluir personal de trabajo social, de apoyo religioso y de ayuda a la infancia para apoyar a la familia (y al niño), cuyo mundo se ha visto por completo trastocado por las palabras "su hijo tiene cáncer".
El tratamiento de los niños con RMS se centra en conseguir un "control local" y un "control sistémico". El control local hace referencia a la erradicación permanente del "tumor primario". Esto se lleva a cabo habitualmente mediante extirpación quirúrgica o irradiación del tumor (o ambos) y de cualquier área cercana afecta, añadiendo además tratamiento quimioterápico. El control sistémico implica la eliminación permanente de las "micrometástasis" invisibles o de las metástasis "visibles", generalmente mediante quimioterapia (a veces con cirugía o radioterapia añadida). El riesgo de que el tratamiento no consiga la curación varía según el "grupo de riesgo". En la mayoría de los niños con tumores no metastásicos (es decir, grupos de riesgo estandar e intermedio), el mayor riesgo estriba en que el tumor primario no pueda ser controlado de manera permanente. Más de la mitad de los fracasos del tratamiento en estos grupos son de tipo "loco-regional" (es decir, en la localización primaria del tumor o cerca de ella). El fracaso en el control del tumor primario se asocia a un notable incremento del riesgo de recaída en otros lugares del organismo; ésto es probablemente un reflejo de la adquisición precoz de resistencia intrínseca a la quimio y radioterapia. En la mayoría de los niños con tumores metastásicos (es decir, de alto riesgo) el riesgo más destacable de fallo del tratamiento estriba en que la metástasis no puedan ser controladas, incluso aunque el tumor primario lo sea. Dado que la supervivencia post-recaída es escasa en la gran mayoría de los niños con RMS recidivante (menos del 20% de los pacientes que recaen se curarán), y aunque existen excepciones, el suministro de un tratamiento óptimo desde el momento del diagnóstico es de importancia crítica.
Tratamiento
El tratamiento de la mayoría de los niños con RMS es administrado bien por grupo cooperativo interdisciplinario o en el ámbito de un ensayo clínico propio de una única institución, bien siguiendo las directrices del ensayo más adecuado. Desde 1972, el grupo de estudio intercentros del RMS (IRSG) ha completado 4 grandes ensayos clínicos prospectivos y secuenciales en los que se trató a unos 4000 pacientes con RMS. En los pacientes con tumores no metastásicos, el estudio más recientemente completado (IRS-IV), plantea dos interrogantes principales derivados de los resultados del análisis estadístico (estudio aleatorio), que son:
1. ¿Mejorará la sustitución de la ciclofosfamida por ifosfamida (VAI) o de la dactinomicina por etopósido (VIE) el pronóstico de los niños con tumores grupo III comparados con la quimioterapia estandar con VAC?
2. ¿Mejorará la radioterapia hiperfraccionada (5940 cGy en dos dosis diarias de 110 cGy) frente a la radioterapia convencional (5040cGy en fracciones diarias de 180 cGy) el control local de la enfermedad?
En pacientes con tumores metastásicos, el ensayo clínico más recientemente completado trató de evaluar la actividad antitumoral y eficacia terapéutica real de uno de los tres pares de fármacos (ifosfamida más doxorrubicina, vincristina más melfalán, e ifosfamida más etopósido) añadidos a la quimioterapia convencional con VAC. osfamide plus etoposide) added to "conventional" VAC chemotherapy.
Los resultados de estos estudios se han ido publicando durante los últimos 7 años.14,34-38 En niños con tumores no metastásicos no se han encontrado diferencias de pronóstico entre las tres formas de tratamiento VIE, VAI, VAC.35 Basándose en ello, la quimioterapia con VAC continua siendo la pauta recomendada por el IRSG como "gold standard" para niños con RMS. Comparándolo con el estudio previo, el IRS-III, el pronóstico mejoró únicamente en un reducido grupo de niños con tumores embrionarios de localización "favorable" no resecados en su totalidad (grupo III) y en aquéllos con tumores resecados bien macroscópicamente, bien en su totalidad (grupos I y II) de localización "desfavorable" (estadios 2 y 3).34 La supervivencia libre de enfermedad a los tres años fue del 77% para el grupo de pacientes; los pacientes con la variante histológica alveolar tuvieron un comportamiento significativamente peor (supervivencia libre de enfermedad a los tres años del 66% frente al 83% de los pacientes con tumores embrionarios). La radioterapia hiperfraccionada no produjo tasas de control local más altas (ni tuvo impacto alguno sobre la supervivencia global) comparada con la radioterapia fraccionada convencional.36 La tasa global de control local de la enfermedad fue del 87%. El riesgo más alto de fracaso local del tratamiento se observó en pacientes con tumores de vejiga o próstata (19%) y parameníngeos (16%).
En pacientes con metástasis, y aunque las tres pautas de fármacos resultan ser muy activas (con tasas de respuesta de entre el 60% y el 80%,37-38 el pronóstico continúa siendo pobre. La supervivencia global del grupo en conjunto es inferior al 30%; se ha sugerido que los pacientes que reciben IE además de VAC tienen mejor pronóstico.38 Se ha observado que el uso de melfalán parece asociarse a una mejoría en la tolerancia a la quimioterapia subsiguiente. No se ha observado beneficio tras consolidación con quimioterapia a altas dosis y rescate con trasplante autólogo de médula ósea.39
Quimioterapia
Todos los pacientes con RMS requieren quimioterapia para aumentar al máximo las posibilidades de curación. La mayoría de los niños en los Estados Unidos son tratados en o siguiendo los preceptos de) un ensayo clínico internacional inicialmente denominado "Estudio Intergrupo del Rabdomiosarcoma" (ahora conocido como Comité de sarcomas de Partes Blandas del Grupo de Oncología Pediátrica). Durante los pasados 30 años se han llevado a cabo 4 Estudios Intergrupo del RMS, con un total de 4000 pacientes tratados. La quinta generación de estos estudios se completará este año. Para pacientes seleccionados, habitualmente aquéllos con RMS de riesgo intermedio o alto, debería existir la opción de tratamiento en el seno de un ensayo clínico piloto institucional.
Los tratamientos quimoterápicos del RMS se administran siempre por vía intravenosa; por lo general se coloca un tipo especial de vía venosa "permanente" antes del inicio del tratamiento. La mayoría de los pacientes con RMS reciben tratamientos quimioterápicos de 6 a 12 meses de duración (raras veces más prolongados, aunque dependiendo de la gravedad de los efectos secundarios el tratamiento diseñado para durar 10 meses puede en ocasiones prolongarse a 15 meses). La quimioterapia se administra por lo general en 2 a 5 (a veces 10) "pulsos" o "ciclos" diarios cada 3-4 semanas. Algunos fármacos quimioterápicos pueden suministrarse semanalmente.
Los efectos secundarios de la quimioterapia pueden ser "específicos" (es decir, que aparecen únicamente con uno o dos fármacos) o "genéricos" (es decir, que se presentan con muchos de ellos). La lista siguiente incluye los fármacos más comunmente utilizados en el tratamiento del RMS en Estados Unidos y en Europa:
- Vincristina
- Dactinomicina
- Ciclofosfamida
- Topotecan
- Irinotecan
- Etopósido
- Ifosfamida
- Doxorrubicina
- Carboplatino
Los efectos secundarios más habituales que pueden aparecer (en mayor o menor grado) con el suministro de la práctica totalidad de los fármacos quimioterápicos usados para tratar el RMS incluyen caída del pelo, náuseas y vómitos, pérdida del apetito, fatiga, llagas o úlceras en la boca y bajos recuentos de células sanguíneas. Estos efectos secundarios aparecen como consecuencia de la acción de los fármacos quimioterápicos sobre las células del organismo que se multiplican con rapidez. Si bien son las células tumorales las que por lo general se reproducen a mayor velocidad, existen otras células normales, como las del pelo, las células que revisten la mucosa oral e intestinal y las células sanguíneas, que también se multiplican en muy poco tiempo. Afortunadamente disponemos de un remanente de dichos tipos celulares superior al de las células tumorales, con lo que los efectos secundarios son habitualmente temporales.
La disminución del recuento de células sanguíneas es el efecto secundario que limita en mayor medida la posibilidad de suministrar quimioterapia de forma continuada y prolongada (tal y como se trataría una infección) y es uno de los más peligrosos. Hay tres tipos principales de células sanguíneas: células rojas (globules rojos o eritrocitos), células blancas (leucocitos) y plaquetas. De forma típica 7 u 8 días después del comienzo de un ciclo de quimioterapia, el recuento de células sanguíneas se sitúa en niveles muy bajos, pudiendo mantenerse disminuido durante 5-10 días. Los glóbulos rojos transportan el oxígeno desde los pulmones al resto del cuerpo; al bajo recuento de glóbulos rojos se denomina anemia, y cursa habitualmente con fatiga.
Los glóbulos blancos son las células del organismo encargadas de la defensa frente a la infección. A la situación de bajos recuentos de glóbulos blancos se denomina leucopenia; esta circunstancia lleva aparejada un gran incremento del riesgo de padecer infecciones graves por gérmenes que están presentes de manera habitual en el organismo. A la situación en la que el tipo de glóbulos blancos más importantes en la lucha frente a la infección está disminuido se la denomina neutropenia.
Las plaquetas son las células que permiten que la sangre se coagule; cuando su recuento es bajo aumenta el riesgo de sangrado, bien espontáneamente o bien tras un corte o traumatismo. Cuando el recuento de glóbulos rojos es bajo, puede realizarse una transfusión que ayude a mejorar la fatiga; también puede suministrarse una transfusión si las plaquetas están bajas, a fin de reducir el riesgo de sangrado. La mayoría de los niños con RMS, incluso aquéllos con riesgo estándar y tumores subgrupo A que reciben quimioterapia no intensiva con 2 fármacos (vincristina y dactinomicina) requerirán soporte transfusional de glóbulos rojos y/o plaquetas en algún momento durante el tratamiento. El único tipo de célula sanguínea que no puede ser trasfundido es el glóbulo blanco destinado a la lucha contra la infección; no obstante, existe un fármaco disponible (G-CSF, filgrastim, Neupogen®) que puede ayudar a los glóbulos blancos a recuperar más rápidamente recuentos que se sitúen en niveles de seguridad.
La inflamación del hígado, aunque es un efecto secundario poco frecuente, puede ocurrir y resultar potencialmente letal, especialmente en niños de corta edad. A fin de detectar de forma precoz su posible aparición es imprescindible una rigurosa y temprana monitorización de los tests de laboratorio que evalúan la función hepática.
La inflamación del hígado, aunque es un efecto secundario poco frecuente, puede ocurrir y resultar potencialmente letal, especialmente en niños de corta edad. A fin de detectar de forma precoz su posible aparición es imprescindible una rigurosa y temprana monitorización de los tests de laboratorio que evalúan la función hepática.
Aunque poco frecuente, el desarrollo de "hepatopatía" quimioinducida puede ser una complicación potencialmente letal. El cuadro se caracteriza por hiperbilirrubinemia, ascitis, coagulopatía e inversión del flujo portal en el estudio ecográfico con Doppler. En niños menores de 3 años el riesgo de padecer esta complicación está muy aumentado. El ajuste de las dosis de fármacos quimioterápicos según la edad del paciente parece reducir el riesgo de hepatopatía, especialmente en niños de corta edad.40
Vincristina es un fármaco suministrado a la práctica totalidad de los niños con RMS. De manera característica puede producir dolor en manos y pies, o en la mandíbula o el abdomen. También causa debilidad en las manos y en los pies secundaria a lesión (habitualmente reversible) en los nervios (cuadro denominado neuropatía periférica). En la actualidad no se dispone de fármacos "protectores" que prevengan el daño de los nervios, pero hay ciertos datos que indican que la lesión nerviosa causada por otros fármacos quimioterápicos no empleados habitualmente en el tratamiento del RMS, en concreto cisplatino y paclitaxel, puede ser minimizado con el uso de vitamina E y de glutamina, respectivamente.
El manejo de la neuropatía periférica por vincristina continúa siendo problemático. Aunque no hay estudios prospectivos rigurosos, la experiencia clínica indica que los pacientes en torno a los 8 años de edad toleran peor el uso intensivo de vincristina que los pacientes más jóvenes. Otros dos fármacos habitualmente empleados en el tratamiento quimioterápico del RMS, cisplatino y paclitaxel, también causan neuropatía periférica. En dos estudios se ha señalado que el uso concurrente de glutamina con el cisplatino,41 y de la vitamina E con el paclitaxel,42 podría reducir la incidencia y severidad de neuropatía periférica. Aunque ningún agente ha sido evaluado de forma reglada en niños con neuropatía periférica asociada a vincristina, existen datos derivados de la experiencia clínica que sugieren que ambos son seguros y bien tolerados, y que pueden ser de ayuda en ciertos casos.
El Irinotecan es un nuevo fármaco que parece ser muy efectivo en el tratamiento del RMS en pacientes diagnosticados de novo con tumores metastásicos y en pacientes con recidiva de RMS (es decir, RMS que reaparecen tras terminar el tratamiento o que tras el tratamiento inicial no desaparecen por completo). Se administra como tratamiento único durante diez días cada tres semanas y, aunque sólo raras veces causa náuseas intensas o vómitos frecuentes, bajos recuentos de células sanguíneas o pérdida del cabello, puede producir cuadros severos de diarrea.
El irinotecan (CPT-11) es un prometedor nuevo fármaco con altas tasas de actividad preclínica en el modelo murino xenografted de RMS.13 Los ensayos clínicos en niños con enfermedad recidivante también demuestran una llamativa actividad.13,43 Los actuales estudios IRS-V lo están utilizando tanto en niños con RMS diagnosticado de novo con metástasis (D9802) como en niños con tumores recidivados (ARST0121). Un ensayo clínico piloto en el MSKCC en pacientes con RMS de riesgo intermedio y alto está también utilizando irinotecan tanto como agente citotóxico "convencional", como potencial agente radiosensibilizante y también como posible agente antiangiogénico cuando se suministra como "terapia de mantenimiento." Aunque es generalmente bien tolerado en lo que respecta a los habituales efectos citotóxicos como alopecia, náuseas y vómitos y citopenias, su uso se relaciona con una alta incidencia de diarrea, incluyendo cuadros severos que requieren rehidratación intravenosa. Se han publicado guías de manejo para esta complicación.44
La ciclofosfamida (habitualmente suministrada en combinación con vincristina y dactinomicina, o vincristina y doxorrubicina) y la ifosfamida (generalmente administrada en combinación con etopósido) pueden causar daño en la vejiga urinaria que cursa con aparición de sangre en la orina. Ambos fármacos han de emplearse con una medicación "protectora" denominada "mesna", eficaz en reducir el riesgo de aparición de este efecto secundario en concreto.
La doxorrubicina puede producir lesión cardíaca, especialmente tras altas dosis totales (acumulativas). Cada vez con mayor frecuencia es administrada, no sólo en casos de RMS sino también en otros tipos de cáncer, junto a un fármaco "de protección" denominado "dexrazoxane", que reduce de forma efectiva el riesgo de esta complicación, potencialmente grave.
A pesar de su destacada actividad antitumoral, el desarrollo de daño cardíaco potencialmente letal, incluso años después de su administración, es uno de los factores que ha llevado a la eliminación de la doxorrubicina de los ensayos clínicos en pacientes con RMS realizados por grupos clínicos cooperativos recientes. Se ha demostrado que el uso de dexrazoxane reduce de forma significativa el riesgo de daño cardíaco asociado a la terapia con doxorrubicina,45 sin detrimento de la efectividad antitumoral de ésta.46
Radioterapia
Todos los pacientes con RMS alveolar- incluso aquéllos cuyos tumores han sido resecados en su totalidad antes del inicio de la quimioterapia- y casi todos los pacientes con RMS embrionario grupo II (con enfermedad residual microscópica) y III (con enfermedad residual visible)- requieren radioterapia para incrementar al máximo las posibilidades de curación. Las niñas con RMS embrionario del tracto genital (vagina, vulva, cérvix y útero) para las que la cirugía conservadora inicial es en buena lógica la regla, pueden con frecuencia ser controladas mediante biopsias seriadas, comenzando aproximadamente 12 semanas después de completado el tratamiento quimioterápico, sin radioterapia adicional, aunque en casos seleccionados (generalmente restringidos a niños con RMS parameníngeo que ha erosionado la base craneal extendiéndose al interior de la bóveda), la radiación debe de comenzar al mismo tiempo (o tan cercana como sea posible) a la quimioterapia.
Dependiendo de la localización, el tamaño y el grupo en que se incluya el tumor, se administran entre 20 y 28 sesiones de radioterapia. De forma óptima el tratamiento ha de planificarse sobre imágenes tridimensionales del tumor de forma previa a la toma de biopsia y al suministro de quimioterapia. Nunca será suficientemente enfatizada la importancia de la habilidad del oncólogo radioterapeuta en el éxito del tratamiento del RMS. Puesto que se trata de tumores poco frecuentes, y dado que la mayoría de los niños con RMS son tratados mediante protocolos en los que se especifica de forma detallada la terapia a seguir en cada caso, el oncólogo radioterapeuta debe de ser capaz no sólo de interpretar con rigor los estudios de imagen relevantes para diseñar un "campo de tratamiento" apropiado que abarque la totalidad del tumor original, añadiendo además un margen adicional de tejido circundante, sino también de llevarlo a cabo en el tiempo especificado en el protocolo y teniendo en cuenta la tolerancia del tejido normal en las estructuras sanas circundantes y los riesgos de complicaciones a largo plazo derivados de la irradiación de tejidos en crecimiento en niños pequeños.
Algunos de los grupos cooperativos europeos que tratan niños con RMS han intentado reducir o suprimir el uso de la radiación en niños muy pequeños, o en niños cuyos tumores han desaparecido por completo después de un período de suministro de quimioterapia, o que se extirparon macroscópicamente por completo antes del inicio de ésta. Desafortunadamente, aunque algunos niños se han curado con este procedimiento, el riesgo de recaída es significativamente mayor y no está claro si la probabilidad de curación es la misma. En consecuencia, y con la excepción de las niñas con RMS embrionario del tracto genital, la radiación está recomendada para todos los pacientes con RMS grupo III, para todos los pacientes con RMS grupo II y para todos aquéllos con RMS alveolar grupo I. El papel de la radioterapia en zonas de enfermedad metastásica en niños con tumores en estadio 4 (o grupo IV) está menos claro, aunque los niños con metástasis pulmonares que han desaparecido tras la quimioterapia podrían tener un mejor pronóstico tras dosis bajas (habitualmente 8 sesiones de tratamiento) de irradiación pulmonar total (WLI).
En el estudio IRS-IV no se han encontrado diferencias entre el uso de radioterapia hiperfraccionada frente a la radioterapia fraccionada convencional (XRT).36 Mientras que la mayoría de los pacientes con tumores en el grupo III conseguirán control local de la enfermedad con radioterapia a dosis completas, aquéllos con afectación ganglionar al diagnóstico se correlacionan con un riesgo 2 veces incrementado de fracaso del tratamiento local.47 Se ha hecho la misma observación en pacientes con tumores pertenecientes al grupo II, en los que el riesgo más alto de recidiva local fue observado en pacientes con enfermedad residual microscópica y afectación ganglionar regional (grupo IIC).48 Todos los pacientes con RMS alveolar, incluso aquéllos con tumores extirpados por completo, deberán recibir irradiación local.49 Los investigadores europeos han tratado de suprimir o limitar el uso de irradiación local en pacientes con tumores en los grupos II50 y III.51 No obstante, con este procedimiento se produjeron tasas de recidiva local significativamente mayores. La importancia de la familiaridad del oncólogo radioterapeuta con las guías de tratamiento de los niños con RMS no debe dejar de enfatizarse nunca.52 El uso del las técnicas de imagen tridimensional y de la radioterapia conformada o modulada por intensidad (nuevas y prometedoras técnicas para suministrar radioterapia altamente dirigida) han conseguido tasas de control local más elevadas, especialmente en pacientes con tumores localizados de alto riesgo o en aquéllos con grandes neoplasias de localización parameníngea con extensión intracraneal.53-54 Unicamente en los pacientes con tumores en el grupo II o III o niñas con tumores embrionarios irresecables del tracto genital, puede no requerirse radioterapia para el control local; el manejo óptimo de estos pacientes consiste habitualmente en una cirugía inicial limitada seguida de biopsias seriadas de control que comienzan aproximadamente tras 12 semanas de tratamiento quimioterápico, con cirugía definitiva o radioterapia 24-30 semanas después si existe persistencia del tumor (la presencia de rabdomioblastos diferenciados no se considera generalmente evidencia de tumor activo en estas localizaciones.55
Cirugía de Rescate (Second-Look)
Algunos niños con RMS serán reintervenidos mediante cirugía de rescate ("second-look") una vez que el tumor se ha reducido de tamaño ("encogido") tras la quimioterapia. Las razones para llevar a cabo este tipo de cirugía incluye el intento de suprimir la necesidad de radioterapia (poco frecuente) o para permitir la aplicación de dosis de radiación post-quirúrgica significativamente más bajas desde el punto de vista clínico (más habitual), así como para maximizar la probabilidad de que la radioterapia post-quirúrgica actúe con mayor efectividad (especialmente en tumores muy grandes en el momento del diagnóstico). En ocasiones, un niño cuyo tumor haya sido tratado con radioterapia puede presentar hallazgos radiológicos preocupantes que sugieran que el tumor no ha sido destruido por la radiación. En estos casos, y si es posible, puede ser necesaria la cirugía para resecar el tumor residual que haya sobrevivido a la radiación y así tratar de prevenir la recidiva del tumor en el lugar primario de origen.
El papel de la cirugía en el manejo de los pacientes con RMS es claramente dependiente y específico de la localización. Se ha sugerido que la práctica de cirugía inicial completa, o con resección macroscópica total del tumor, o incluso de reducción de masa tumoral, en pacientes con tumores de localización desfavorable, se asocia a un mejor pronóstico.56-58 Dado que la realización de un estudio aleatorio de resección quirúrgica es muy improbable, con casi total seguridad nunca será posible determinar si la mejoría en el pronóstico es resultado de la cirugía per se, o si la susceptibilidad de resección quirúrgica se asocia simplemente con otros factores relacionados con un mejor pronóstico, como son la presencia de tumor residual macroscópicamente visible en la reexploración pretratamiento en pacientes en los que se presuponía resección inicial "completa", los tumores de pequeño tamaño o no invasivos, aquéllos con ausencia de afectación ganglionar y en tumores con mejor respuesta a la quimioterapia neoadyuvante. Como norma general, especialmente en pacientes con tumores de localización desfavorable, lo más adecuado es realizar en el momento del diagnóstico una cirugía que conserve al máximo la función y el aspecto estético. Para tumores que no pueden ser resecados al diagnóstico, deberá considerarse la posibilidad de cirugía de recate, especialmente si es factible la extirpación total o macroscópica completa que permita una reducción significativa de la dosis de radioterapia post-operatoria, o bien si se sospecha la presencia de tumor residual viable tras la radioterapia.59 Aunque la cirugía "no mutilante" ha sido el principio guía en las dos décadas pasadas, particularmente en pacientes con tumores de vejiga o próstata, un estudio reciente ha destacado la prudente puntualización de que la conservación del órgano no equivale necesariamente a conservación de la función normal de éste.60
Nuevos Tratamientos
La supervivencia post-recaída de la mayoría de los pacientes con RMS recidivado continúa siendo desalentadora. El 95% de las recidivas tienen lugar dentro de los 3 años posteriores al diagnóstico. Con la excepción de un pequeño grupo de riesgo favorable (aproximadamente el 20% de los pacientes con recidiva del tumor), cuya supervivencia a los 5 años se aproxima al 50%, la mitad de los pacientes con RMS recurrente fallecerán por su enfermedad durante el primer año tras la aparición de la recidiva y el 90% en los 5 años tras ésta.61
Para este grupo de pacientes se necesitan de forma acuciante nuevas terapias.
A medida que se consigue profundizar en los procesos críticos de la "rabdomiosarcomagénesis,"62-64 se abren nuevas vías para el desarrollo de tratamientos basados en la biología tumoral. Los tratamientos dirigidos a interrumpir las interacciones clave entre el receptor de un factor de crecimiento y su ligando, o de los productos de su acción en cascada, parecen ser particularmente prometedores. La ruta autocrina del IGF-II juega un papel en el crecimiento del RMS;65 la interrupción de esta vía constituye una atractiva terapia biológica potencialmente utilizable. El crecimiento de xenoinjertos de RMS en ratones puede ser inhibido con el uso de anticuerpos monoclonales dirigidos contra el receptor de IGF-I, que se une a IGF-II y media su señal mitógena.66 Un nuevo anticuerpo monoclonal que reconoce el receptor humano de IGF-I ha demostrado ser capaz de inhibir la proliferación inducida por IGF-I en una línea celular de RMS.67 Se han sintetizado pequeñas moléculas altamente específicas con acción inhibidora de la tirosín-kinasa dirigidas contra el receptor tirosín-kinasa IGF-I; estas moléculas han demostrado ser capaces de inhibir el crecimiento de los xenoinjertos tumorales, tanto por sí solos como en combinación con quimioterápicos citotóxicos.68
El conocimiento de que las proteínas intracelulares pueden ser procesadas y presentadas por el complejo mayor de histocompatibilidad (MHC) como péptidos en la superficie celular ha llevado a sugerir la posibilidad de que los productos de ciertos genes mutantes específicos de tumor pudieran ser la diana de las células T citotóxicas.69-70 Los investigadores han demostrado que un péptido derivado de una forma mutante de la proteína p53 es reconocida de forma específica por las células T citotóxicas.71-72 De manera análoga, las proteínas de fusión específicas de traslocación pueden ser la diana de las células T citotóxicas (CTL). Más específicamente, la proteína de fusión PAX-FKHR generada por la traslocación t(2;13)(q35;q14) en el RMS alveolar es una diana potencial en la terapia con CTL. En estudios preclínicos en murinos se ha demostrado que las células dendríticas derivadas de la médula ósea (DCs) estimuladas con Antígenos asociados a Tumor (TAA) pueden dar lugar tanto a células Natural Killer (NK) como a linfocitos T-citotóxicos CD8+ frente al RMS.73 Basándose en este descubrimiento, se están desarrollando estudios clínicos piloto que usan vacunas elaboradas con péptidos específicos PAX-FKHR generados en células dendríticas. El éxito de este avance dependerá de la capacidad de las células tumorales para presentar un péptido de fusión procesado y unido al MHC en la superficie celular. Si ésto es factible, se podrán realizar múltiples ensayos destinados a superar los potenciales déficits que permiten al tumor escapar inicialmente de la inmunidad.74-75
A medida que se consiguen mayores avances en el conocimiento de la biología básica del RMS, se van desarrollando aproximaciones al diseño de tratamientos novedosos que tratan de sacar ventaja de este "tendón de Aquiles" de las células tumorales. Dada la dependencia del RMS del IGF-II, se están desarrollando nuevos y prometedores fármacos que actúan ya sea bloqueando la interacción entre IGF-II y el receptor de IGF tipo I o bien la secuencia de efectos biológicos en cascada que tienen lugar tras la unión de IGF-II a su receptor. Estos agentes, aunque aún no disponibles para uso clínico, son muy prometedores tanto como tratamiento único como en combinación con quimioterapia.
Finalmente, dada la presencia de un gen específico de célula tumoral, derivado de una traslocación, en los casos de RMS alveolar, existe la posibilidad de utilizar terapias inmunológicas que reconozcan y destruyan aquéllas células que contienen el gen anormal. Se están llevando a cabo ensayos clínicos piloto que evalúan la capacidad de pacientes con RMS alveolar "vacunados" para desarrollar inmunidad frente a sus propios tumores; de forma simultánea se están poniendo también en práctica ensayos clínicos piloto destinados a evaluar la capacidad del sistema inmunitario de un familiar "genéticamente compatible" para controlar el RMS alveolar de un paciente tras un trasplante "mini" alogénico de células madre.
Efectos a Largo Plazo del Tratamiento del Rabdomiosarcoma
La implantación de terapias ajustadas al riesgo en los niños con RMS se basa en el intento de maximizar la probabilidad de curación reduciendo simultáneamente la aparición de complicaciones a corto, medio y largo plazo. Los efectos tardíos relacionados o atribuibles a la terapia pueden desarrollarse meses o incluso años después de haberse completado el tratamiento. Los agentes quimioterápicos pueden, de forma individual, tener un efecto tóxico particular y específico que no se manifieste hasta muchos años después de la terminación de la terapia, o que empeore de forma continua a medida que se alarga el tiempo de seguimiento. Las lesiones por radioterapia, así como las complicaciones tardías tras la cirugía, pueden no ser aparentes durante muchos años, especialmente en niños en crecimiento. Algunas complicaciones concretas bien conocidas del tratamiento son:
- Infertilidad (asociada especialmente con el uso de agentes alquilantes como la ciclofosfamida y la ifosfamida): El riesgo de aparición de infertilidad inducida por quimioterapia es mucho mayor en los niños que en las niñas.76 Siempre y cuando sea factible, incluso en varones en el pico de crecimiento puberal, debe valorarse la posibilidad de criopreservación de esperma.77 Pese a que son objeto de intensa investigación en los laboratorios, la criopreservación del tejido ovárico o la de las ovogonias no es actualmente un procedimiento factible como técnica rutinaria de preservación de la fertilidad en niñas;78 afortunadamente, el riesgo de infertilidad parece ser mucho más bajo en éstas que en los varones. Para aquéllas que vayan a recibir irradiación pélvica, o para niños a los que vaya a suministrarse irradiación escrotal, la trasposición quirúrgica de la(s) gónada(s) lejos del campo de radiación puede ser de ayuda en la preservación de la función hormonal y/o de la fertilidad.
- Disfunción vesical: Aunque la cirugía conservadora "no mutilante" y la irradiación a dosis completa se ha convertido en el tratamiento de elección para la preservación de la vejiga en niños con RMS prostático o vesical, aproximadamente la mitad de los niños con vejigas "intactas" tendrán uno o varios de los síntomas propios de la disfunción vesical, incluyendo goteo miccional, incontinencia urinaria y enuresis.60
- Daño por radiación de las estructuras de cabeza y cuello: El uso de la radiación para tratar tumores originados en las estructuras de la cabeza y el cuello es con frecuencia impracticable dada la ausencia de órganos o tejidos "no esenciales" que puedan ser "sacrificados" si se intenta realizar una resección quirúrgica completa. Entre las complicaciones bien conocidas de la radiación se incluyen la formación de cataratas tras dosis del globo ocular tan bajas como de 1000 cGy;79 el desarrollo facial asimétrico como resultado del crecimiento de hueso atrapado de forma permanente y de la fibrosis ("cicatrización") de los tejidos circundantes; infecciones crónicas de los senos paranasales; retraso del crecimiento secundario a lesión hipofisaria;80 y anomalías dentarias complejas múltiples.81 Se desconoce si ciertas técnicas nuevas de radioterapia, dirigidas de forma más precisa, como la Radioterapia de Intensidad Modulada (IMRT), reducirá el riesgo de complicaciones tardías derivadas de la irradiación de las estructuras de cabeza y cuello.53
- Neoplasias secundarias (SMN): Quizá la complicación tardía más devastadora del tratamiento de cualquier tipo de cáncer, y no sólo del RMS, sea el desarrollo de una segunda neoplasia maligna (SMN). El uso de radio y quimioterapia pueden llevar al desarrollo de segundas neoplasias malignas. Las neoplasias secundarias asociadas a quimioterapia son más frecuentemente leucemias (típicamente leucemias mieloides agudas [AML]) y podrían estar relacionadas con el uso de agentes alquilantes (ciclofosfamida e ifosfamida) y de inhibidores de la topoisomerasa II (etopósido y doxorrubicina). El riesgo de aparición de una leucemia secundaria es, afortunadamente, bastante bajo (generalmente de entre el 1 y el 2%). La radiación también se asocia al desarrollo de cánceres secundarios, más frecuentemente otros sarcomas (ya sea del hueso o de partes blandas). A las dosis que se usan habitualmente para tratar niños con RMS, el riesgo de desarrollo de sarcomas secundarios es aproximadamente del 5% a los 20 años.82 A diferencia de lo que sucede con las leucemias secundarias, que de modo característico aparecen en los primeros 4 años tras el tratamiento, la mayoría de los sarcomas secundarios no se desarrolla hasta pasados 5 años o más del fin de éste.82 La posible contribución de "factores de riesgo genético" subyacentes en el desarrollo de las neoplasias malignas inducidas por el tratamiento está siendo investigada de forma activa.
En los 1770 pacientes incluidos en los estudios IRS-I e IRS-II aparecieron veintidós neoplasias malignas secundarias, que incluían 11 casos de sarcomas en relación con la radiación y 5 casos de leucemia aguda no linfoblástica, aparecidos tras una media de 7 años post-tratamiento.83 Tres de los pacientes afectados padecían neurofibromatosis, y las familias de otros siete presentaban historia compatible con LFS; ésto sugiere que la susceptibilidad genética juega un papel significativo en el desarrollo de una neoplasia maligna secundaria tras el tratamiento del RMS. Los resultados iniciales del estudio IRS-III describen la aparición temprana de 5 casos de leucemia mieloide aguda en niños, así como la de un caso de osteosarcoma y otro caso de síndrome mielodisplásico.84 Los resultados preliminares del estudio IRS-IV en relación a las SMN incluyen la aparición de 14 casos en 13 pacientes en periodos cuya media es de 3.2 años desde el diagnóstico.85 Una más reciente actualización actualización de los resultados del IRS registra 67 de SMN y 2 casos de tercera neoplasia maligna en un total de 4367 pacientes incluidos en los estudios IRS entre los años 1972 y 1997.86 Sólo 7 tenían un síndrome de predisposición genética conocido. La incidencia acumulada estimada de SMN a los 20 años fue del 3.5%. Los hallazgos iniciales en relación a un posible riesgo incrementado de AML/MDS en pacientes que reciben etopósido no parecen sustentar esta hipótesis; no obstante, resultaría prudente realizar una monitorización prospectiva de la posible contribución de una fuerte historia familiar de cáncer en la evaluación del riesgo de desarrollo de una SMN relacionada con el tratamiento.87
-
Aproximadamente dos tercios de los niños con RMS tienen menos de 10 años en el momento del diagnóstico. El RMS es más frecuente en niños de entre 1 y 4 años de edad, y muy raro en menores de un año.
-
La RMN de la órbita muestra una masa de partes blandas originada en la cara anteromedial de la órbita izquierda que desplaza hacia adelante y lateralmente el globo ocular.
-
La RMN de los senos paranasales muestra una gran masa de partes blandas centrada en la region sinonasal que invade ambas órbitas y se extiende al interior del cráneo a través de la base craneal.
-
La imagen corresponde a una RMN prostática y muestra una gran masa de partes blandas en el lado derecho de la pelvis que comprime la pared posterior de la vejiga urinaria y la pared anterior del recto.
-
El RMS embrionario es de forma característica menos denso y más fusiforme. La diferenciación muscular estriada de esta variante puede ser evidente o no en el examen histológico rutinario. La confirmación de la naturaleza muscular esquelética del tumor mediante técnicas inmunohistoquímicas, con el uso de anticuerpos específicos para desmina, vimentina, actina y miogenina, apoyan el diagnóstico de RMS.
-
Las células del RMS alveolar son típicamente más pequeñas y redondas y dan una apariencia más densamente celular al tumor. Arquitecturalmente se distribuyen revistiendo pseudoespacios que remedan los pequeños espacios aéreos saculares distales del pulmón (alveolos).
-
Figura 8: Sobreexpresión del factor de Crecimiento análogo a la Insulina tipo II (IGF-II) por pérdida de heterocigosidad en 11p15.
-
Figura 9: La translocación recíproca entre PAX y FKHR crea un "oncogén híbrido"









{kind=link}
{kind=link}
{kind=link}
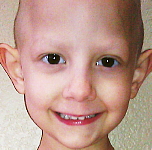{kind=link}
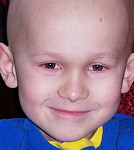{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}